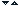Disease
|
Enzyme Deficiency
|
Major Accumulating Metabolites
|
4. Corpusculii Dutcher
5. corpusculii Lewy
a1-antitripsina
V. ACUMULARI INTRACELULARE DE PIGMENTI
TULBURARILE PIGMENTULUI MELANIC
Melanocitele: localizare
origine - migrare din
creasta neurala
Biologie:
melanina - se formeaza in melanocite: oxidarea tirozinei in
dihidroxifenilalanina (DOPA) sub actiunea tirozinazei
-
sinteza: in melanozomi
-
transfer in: cheratinocite, celulele corticalei firului de par
TULBURARI CU MINUS
DE MELANINA (HIPOMELANOZE)
Hipomelanoze congenitale
Albinismul
Albinismul oculocutanat
AR
Defect: mutatii ale genei
tirozinazei (11q) inactivitate
ME - melanocite prezente
Clinic: piele
par
pupile
Complicatii: fotofobie, tulburari de vedere
cheratoza
actinica
cancere
cutanate
Albinismul ocular
transmitere X-linkata
Piebaldismul
("albinism partial", leucodermie "ornamentala")
AD
Microscopie: absenta melanocitelor (lipsa genei ce
codifica migrarea)
Clinic:
de la nastere
nu se
modifica
suprafata
ventrala, central (scalp, frunte, fata, trunchi,
extremitati)
Sindromul Chediak-Higashi
AR
Patogeneza:
anomalii ale
granulelor lizozomale din diferite celule (melanocite, neutrofile,
fibroblaste etc.)
consecinte:
- functia neutrofilelor
- functia melanocitelor
Clinic: piele galbuie,
par blond, iris palid
infectii
Hipomelanoze dobandite
Vitiligo
Clinic
- arii
depigmentate
- halou
- pilozitatea
pastrata
- localizare
Microscopie: distrugerea melanocitelor
Patogeneza: autoimuna; asociere
cu alte boli autoimune, Li T
Postinflamatorii: sarcoidoza
cutanata, lepra
Depigmentari
chimice:
hidrochinone, fenoli
Copyright © 2025 - Toate drepturile rezervate
|