|







 |  |
|
| |
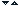 | Aeronautica | Comunicatii | Constructii | Electronica | Navigatie | Pompieri |
| Tehnica mecanica |
Catalizatorii oxizi semiconductori suportati
Catalizatorii, in particular cei pe baza de oxizi, sunt supusi unor transformari in timpul actului catalitic, aceste transformari fiind traduse in restructurari ale suprafetei (uneori ireversibile), datorate adesea de interactia cu reactantii, dar si cauzate de procese paralele cum ar fi deshidratarea, hidratarea sau reactia in faza solida intre componente [30]. De aceea in timpul reactiei se pot inregistra schimbari pozitive ale activitatii catalitice (cum ar fi binecunoscuta activare a catalizatorilor de oxidare selectiva prin reducere partiala), dar si fenomene nedorite de dezactivare.
De obicei suprafata catalizatorului atinge un nivel stabil de activitate dupa un tratament preliminar, care o "formateaza", creand centrii activi capabili de a determina desfasurarea reactiei in directia dorita. Distributia finala a produsilor este influentata de calitatea acestei "formatari". In acelasi timp, stabilitatea suprafetei active depinde de masura in care excesul sau deficitul de sarcini rezultate din procesele de transfer de sarcini cu reactantii sunt reechilibrate de reteaua solidului, suprafata fiind in permanenta intr-un echilibru dinamic nu numai cu reactantii gazosi, ci si cu interiorul ("bulk"-ul) solidului. Deci controlul eficient al unui proces catalitic necesita informatii despre dinamica interactiei intre reactanti si catalizator, in particular in legatura cu procesele de transfer de sarcina la suprafata.
Un fenomen interesant observat in unele reactii este acela de crestere a activitatii catalitice prin amestecarea mai multor faze sau prin depunerea pe un suport. Acest efect sinergetic (care are ca rezultat cresterea capacitatii de adsorbtie a sistemelor polifazice ca si activarea unei faze in prezenta alteia) a fost explicat uneori prin transferul de specii intre faze (efectul de spillover) [31]. Mecanismul acestui proces nu a fost inca suficient explicat in ciuda numeroaselor date experimentale si a interesului major de a cunoaste si exploata in practica aceste fenomene intilnite adesea si in cazul catalizatorilor suportati.
Folosirea suportilor este o practica curenta in prepararea catalizatorilor, deoarece reprezinta o modalitate de a rezolva multe probleme de interes practic si anume:
-obtinerea unor catalizatori cu un pret de cost mai scazut (prin utilizarea unei cantitati mai mici de faza activa)
-marirea suprafetei specifice si deci a eficientei actului catalitic.
-stabilizarea unor faze active intr-o anumita structura dorita.
Considerat initial ca inert din punctul de vedere al reactiei, practica a dovedit ca suportul joaca de fapt un rol important in structurarea catalizatorului final, mai ales in ceea ce priveste proprietatile acido-bazice ale suprafetei active. Desi se stie ca zonele de contact intre faza activa si suport functioneaza ca o interfata pentru procese de transfer de specii si/sau sarcini electrice intre cele doua solide, acest fenomen nu este inca studiat sistematic, alegerea suportilor fiind dictata de criterii empirice. Intelegerea mecanismelor complexe de implicare a suportului in controlul structurii/activitatii fazelor active este de mare importanta, pentru obtinerea unor catalizatori cu proprietati pre-definite.
Un numar mare de oxizi sunt catalitic activi in reactii de oxidare. Desi utilizarea catalizatorilor oxizi este limitata de comportarea lor variabila si instabilitatea structurala la temperatura ridicata in conditii reducatoare, folosirea lor pentru combustie ar avea avantajul pretului de cost substantial mai coborat, cu atat mai mult cu cat unii dintre oxizi sunt si catalizatori foarte eficienti pentru oxidarea CO [32-34] sau pentru reducerea NOx [35]. Extinderea in ultimii ani a aplicarii materialelor anorganice in moduri tot mai sofisticate in electronica, cataliza si la obtinerea unor materiale ceramice speciale a determinat si concentrarea cercetatorilor pe studierea si punerea la punct a unor metode de preparare a acestor materiale in conditii care sa permita controlul foarte fin al proprietatile lor fizico-chimice, in special cand este vorba de amestecuri de oxizi, sau de faze active depuse pe suport.
Depunerea unui compus anorganic pe un suport necesita indeplinirea simultana a mai multor conditii. Dintre acestea, una din cele mai importante o constituie realizarea unei depuneri stabile, care rezulta din compatibilitatea suportului cu precursorul fazei depuse. De aceea, metoda folosita pentru obtinerea catalizatorului suportat trebuie sa aiba in vedere alegerea unui precursor cat mai adecvat, cu o structura cat mai omogena, care sa poata fi obtinut in mod reproductibil, si la cea mai joasa temperatura posibila, pentru a mentine pe cat posibil o microporozitate adecvata cat si o suprafata specifica initiala a suportului cat mai putin alterata.
In literatura se cunosc in prezent cateva tipuri mari de metode folosite pentru obtinerea de materiale suportate si anume:
metoda impregnarii,
metoda coprecipitarii,
metoda sol-gel
metoda amestecurilor mecanice de oxizi.
Dintre acestea, metoda impregnarii suportului cu sare solubila a cationului al carui oxid metalic dorim sa-l depunem este inca metoda cea mai utilizata, datorita avantajelor sale majore, cum ar fi aceea ca permite obtinerea unor incarcari foarte diverse, prin modificarea concentratiei solutiei de impregnare .
In acelasi timp, pentru a se asigura o depunere a oxidului metalic pe suport "rezistenta" mecanica si stabila la manipularile de rutina, este absolut necesar ca in fazele de preparare sa existe o afinitate structurala intre suport si precursorul fazei oxidice active
I.2.2. Dioxidul de staniu (SnO2). Generalitati.
Un caz mai special este reprezentat de oxizii cu proprietati catalitice si in acelasi timp semiconductoare (de exemplu SnO2, MoO3, V2O5); aceasta combinatie de proprietati sta la baza aplicarii lor si ca senzori catalitici, folositi la monitorizarea compozitiei unor gaze. Unii dintre oxizii de acest tip (cum ar fi SnO2) ar putea fi folositi pentru obtinerea unui catalizator care sa indeplineasca simultan mai multe functiuni:
catalizator de combustie
catalizator de depoluare
senzor catalitic
In ciuda faptului ca activitatea sa catalitica intrinseca este neimportanta, dioxidul de staniu este foarte mult utilizat in practica, la obtinerea de catalizatori industriali pentru oxidarea si oxi-dehidrogenarea selectiva sau pentru izomerizarea hidrocarburilor [36-39], ca un constituent de baza al senzorilor de gaze [40-42] sau drept material de electrod [43], in particular datorita proprietatilor sale de semiconductor de tip n ca si a stabilitatii sale structurale.
Oxidarea catalitica a hidrocarburilor, hidrogenului sau a monoxidului de carbon pe dioxid de staniu se desfasoara printr-un mecanism clasic de tip redox (Mars-van Krevelen [44]):
Reducerea catalizatorului oxidat
![]()
Oxidarea catalizatorului redus
![]() .
.
In functie de conditiile experimentului (temperatura de lucru, concentratie de reactanti), produsii obtinuti la oxidarea hidrocarburilor pe SnO2 sunt fie compusi de oxi-dehidrogenare, (la temperaturi mai coborate), fie produsi de oxidare totala, (peste 350oC) [45].
Inconvenientul principal pentru posibila aplicare a oxidului de staniu drept catalizator de depoluare/combustie il constituie suprafata sa specifica coborata (maximum 10 m2/g in mod uzual), ceea ce limiteaza considerabil eficienta actiunii sale. O posibilitate de ameliorare a acestui inconvenient este dispersarea sa prin depunerea pe suporti de suprafata specifica mare, cum ar fi alumina, dioxidul de titan sau dioxidul de silice.
Una din problemele ridicate de senzorul cu SnO2, valabila de altfel si in cazul celorlalti senzori, este selectivitatea lui coborata, adica faptul ca raspunde la un spectru larg de gaze reducatoare. De asemenea dioxidul de staniu are o comportare variabila datorata sensibilitatii la umiditatea atmosferica. Motivele acestei comportari sunt numai in parte cunoscute sau explicate [46,47]. S-a aratat [48] ca in prezenta umiditatii la temperatura joasa este impiedicata adsorbtia de oxigen, deoarece apa concureaza cu acesta pentru adsorbtia pe aceeasi centri.
In domeniul de temperatura 200-1350oC, Miusaki [55], a pus in evidenta in SnO2, existenta unui deficit de oxigen fata de compozitia stoechiometrica. Densitatea golurilor (definite prin x in formula SnO2-x), depinde puternic de temperatura si de presiunea partiala de oxigen. Acestea variaza in general intre 10-3 si 10-2 % at.
Structura cristalografica si electronica a SnO2
Structura dioxidului de staniu este de tip rutil (Figura). Celula elementara este tetragonala (a=b=0,475 nm, c=0,318nm) si contine sase atomi: doi atomi de staniu si patru atomi de oxigen. Fiecare atom de de staniu este centrul unui octaedru aproape regulat format din ssase atomi de oxigen, in timp ce fiecare atom de oxigen este inconjurat de trei atomi de staniu situati pe varfurile unui triunghi isoscel.
Razele ionice ale cationului Sn4+ si ale anionului O2- sunt de 0,0071 si 0,14 nm.
Structura electronica
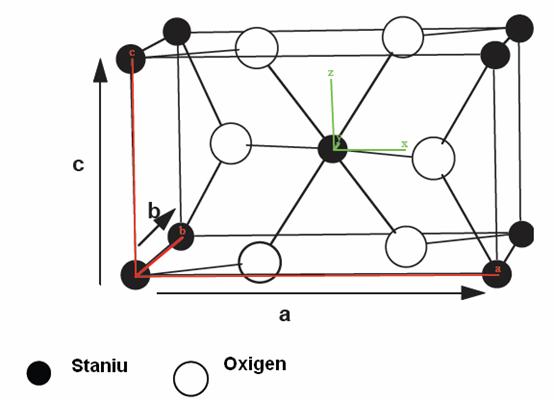
Oxidul de staniu este un semiconductor cu o banda interzisa de 3,6 eV [56, 57]. Vacantele de oxigen formate prin transferul atomului de oxigen in stare gazoasa, permit obtinerea unui semiconductor de tip n. Intr-adevar, vacanta de oxigen astfel creata poseda 2 electroni, care pot fi cedati la cresterea temperaturii. Electronii liberi se pot atasa de atomii de Sn4+, care devin astfel Sn2+ si se comporta ca si donori de electroni.
Diagrama energetica a SnO2 dupa 86]:
Rolul suportului ca sursa/consumator de purtatori
Reactiile catalitice heterogene folosesc drept catalizator un numar foarte mare de solide (metale, oxizi, sulfuri, etc) ce au proprietatea speciala de a facilita in mod specific transformarea reactanttilor intr-o directie dorita. O mare parte din acesti compusi (numiti in mod curent faze active) prezinta inconvenientul unor suprafete specifice mici, ceea ce limiteaza substantial eficienta lor in reactie.
Depunerea pe suporti de suprafata catalitica mare a constituit solutia cea mai simpla, pornindu-se initial de la ideea ca acesti suporti ar fi inerti pentru reactia studiata, urmand a juca doar rolul unei faze de dispersie. Ulterior, s-a constatat ca in multe situatii suportul are o contributie proprie la realizarea propietatilor catalizatorului final. In ciuda faptului ca prepararea stiintifica a catalizatorilor este un domeniu deja bine definit al catalizei, alegerea suportilor se face inca in mod uzual dupa criterii destul de empirice, iar natura influentei lor este inca insuficient studiata.
Oxidul de aluminiu exista in cateva modificari structurale, dintre care faza g este cel mai mult utilizata atat drept catalizator, cat si ca suport catalitic [58-61]. Tranzitia intre diferitele faze inrudite este determinata de deshidratarea progresiva, forma a fiind forma de temperatura inalta si cea mai stabila din punct de vedere structural [61]. Avantajul esential al aluminelor de tranzitie folosite in cataliza este reprezentat de suprafata lor specifica foarte mare (intre 100-200m2/g) stabila in conditiile de exploatare curenta a catalizatorilor in cauza. Deoarece inca de foarte mult timp s-a dovedit ca proprietatile g-Al2O3 depind in mare masura de gradul sau de deshidratare, un numar de studii s-au dedicat in mod specific urmaririi capacitatii de adsorbtie a apei ca si a stabilitatii gruparilor OH pe suprafata g-aluminei. [58,61-63]. Aceste studii au fost determinate in particular de faptul ca proprietatile catalitice ale g-Al2O3 sunt atribuite prezentei pe suprafata a centrilor acizi de tip Lewis, ce se formeaza prin indepartarea gruparilor OH superficiale datorita calcinarii la temperaturi peste 400oC.
Pe de alta parte insa, alumina adsoarbe foarte usor apa, iar adsorbtia de apa (intervenita chiar si accidental, in timpul manipularilor), poate determina modificarea radicala a distributiei centrilor acizi, schimband balanta intre centrii acizi de tip Lewis si cei de tip Brønsted. Distinctia intre diferitele tipuri de adsorbtie a apei - fizica, ca specie moleculara sau adsorbtie chimica nu este inca foarte precisa, deoarece criteriile alese pentru definirea lor au fost partial empirice. In general, strategia abordata pentru evaluarea continutului total de apa/grupari OH in toate aceste lucrari a fost aceea a deshidratarii avansate (prin evacuarea la vid, la incalzire) urmata de re-hidratare si controlul cantitatilor de apa retinute sau pierdute la anumite temperaturi. Insa, in ciuda precautiilor luate, rezultatele sunt grevate de un anume grad de incertitudine, deoarece:
g alumina contine o anumita cantitate de apa in structura sa. Indepartarea sa "totala" prin evacuare si incalzire la o temperatura moderata este dificila, deoarece o parte din apa (apa "din capilare") este inglobata in spatiile dintre cristalite sau in zonele de contact intergranular in cursul procesului de obtinere a materialului; se presupune ca gruparile OH prezente pe muchiile/colturile cristalitelor faciliteaza de asemenea adsorbtia apei moleculare prin punti de hidrogen. Acest tip de apa se elimina greu, prin difuzia lenta a speciilor catre suprafata, deci durata implicataa in operatia de eliminare a apei moleculare este importanta. De aceea, este dificil sa se faca cu certitudine evaluarea raportului dintre apa readsorbita si apa "interna" retinuta in retea ca urmare a tratamentelor preliminare.
Incalzirea si evacuarea la o temperatura mai ridicata (pentru accelerarea procesului de deshidratare primara) risca sa provoace dehidroxilarea si chiar tranzitia ireversibila a fazei g catre faza a, ceea ce schimba total informatiile despre gruparile OH superficiale si modifica ireversibil suprafata. Pe de alta parte, in cursul incalzirii se poate petrece si un proces de chemosorbtie, parte din apa ramasa in solid reactionand cu suprafata.
Limita de temperatura la care se considera ca are loc indepartarea apei adsorbite fizic a fost aleasa arbitrar ca fiind intre 100-120oC [62-65]. Pe baza faptului ca un numar de grupari OH (aparute prin adsorbtia apei la temperatura camerei pe aluminele cu grad avansat de deshidratare) persista pe suprafata prin evacuarea la 120oC, s-a dedus ca apa se poate chemosorbi chiar si la temperatura camerei [64]; masuratorile prin spectroscopie in IR au aratat insa ca pentru chemosorbtia apei e nevoie de incalzire [63], si temperaturi de chemosorbtie de 100 sau 300oC sunt considerate in alte lucrari [64, 65].
Temperatura la care incepe desorbtia apei chemosorbite reprezinta de asemenea o problema importanta pentru conditionarea optima a materialelor oxidice in particular pentru masuratori spectroscopice; au fost folosite diverse tehnici pentru identificarea acestei temperaturi.
Masurarea caldurilor de imersie [66] a dovedit ca, asa cum era de asteptat, stabilitatea termica a gruparilor OH superficiale in diversi oxizi depinde de natura cationului. S-a stabilit ca dehidroxilarea incepe la 110oC pentru TiO2, 200oC pentru ZnO [66] si peste 2500C pentru SnO2 [67]. Pentru g-alumina se considera ca aceasta temperatura este de 3000C [59], dar aceasta valoare a fost de asemenea stabilita arbitrar.
Se pune in mod necesar problema de a se face o distinctie clara intre apa adsorbita fizic si apa chemosorbita, si mai ales de a se stabili in ce masura aceste specii coexista in timpul reactiilor catalitice ce se desfatoara la temperaturi mai coborate, avand in vedere ca in procesele catalitice curente, nu se aplica proceduri de tipul evacuarii la vid inalt. Suprafetele catalizatorilor sunt adesea contaminate cu contaminanti atmosferici, apa fiind unul dintre cei mai importanti.
In masura in care pentru reactia catalitica se folosesc domenii de temperatura foarte diferite, raportul intre centrii acizi de tip Brønsted si Lewis poate fi mult diferit de cel asteptat pe baza rezultatelor obtinute prin experimente "model".
Masuratorile de conductibilitate electrica pe probe oxidice policristaline sunt foarte sensibile la prezenta apei pe suprafata sau in zonele intergranulare si pot fi utilizate, prin masuratori "in situ" la studiul etapelor de hidratare/deshidratare la presiunea atmosferica si in curent de gaz.
In lucrari efectuate in colectivul nostru [68, 69] s-a aratat ca masuratorile de conductibilitate electrica in curent alternativ, cand sunt efectuate in situ pot fi folosite pentru urmarirea dinamicii suprafetelor catalitice in cursul unor procese de deshidratare respectiv hidratare si pentru identificarea stabilitatii termice a unor specii adsorbite pe suprafata. In particular in cazul proceselor de hidratare sau deshidratare efectul produs asupra conductibilitatii electrice este datorat atat reorientarii unor dipoli (cum ar fi chiar moleculele de apa si gruparile OH in prezenta campului electric), cat si modificarii raportului intre conductia de tip protonic si cea prin purtatori electronici. Intr-adevar, este cunoscut faptul ca toti oxizii contin mai mult sau mai putin grupari OH superficiale, deoarece acestea fiind mai polarizabile decat ionii de oxigen ai retelei, pot contribui mai eficient la minimalizarea neregularitatilor energetice ale suprafetei. In masura in care parte din aceste grupari sunt polarizate astfel incat protonul aferent este relativ mobil, oxizii pot prezenta conductie protonica, protonul deplasandu-se in mod esential prin doua tipuri de mecanisme: mecanismul de tip vehicul (specia mobila fiind H3O+ sau NH4+) sau prin mecanism de tip Grotthuss (salt ionic intre grupari OH invecinate) [70]. In conditii favorabile prezentei pe suprafata a apei moleculare, are loc transferul prin mecanismul vehicul. Pierderea apei prin fluxare/incalzire blanda determina scaderea dramatica a moleculelor mobile si trecerea catre conductia prin mecanism Grotthuss. Pe baza analizei simultane a parametrilor de conductie (conductia G, capacitatea C si energiile de activare) ca si a compusilor existenti pe suprafata in diferite domenii de temperatura (prin folosirea unei metode analitice asociata) se pot identifica domeniile de stabilitate termica pentru diferite specii superficiale si se pot obtine informatii indirecte privind natura suprafetei intr-un interval de temperatura de interes.
Gamma alumina
Oxidul de aluminiu exista in cateva modificari structurale (dintre care siAl2O3 sunt cele mai utilizate drept catalizator, cat si ca suport catalitic). Tranzitia intre diferitele faze inrudite este determinata de deshidratarea progresiva, forma fiind forma de temperatura inalta si cea mai stabila din punct de vedere structural. Avantajul esential al aluminelor de tranzitie folosite in cataliza este reprezentat de suprafata lor specifica foarte mare (pana la 300 m2/g pentru -Al2O3), relativ stabila in conditiile de exploatare curenta a catalizatorilor in cauza. In particular alumina este foarte mult utilizata atat ca suport, cat si drept catalizator in multe procese industriale, in special in petrochimie. De aceea alumina, si toata seria aluminelor de tranzitie au facut obiectul unor studii detaliate studiul [71-74], care se focalizeaza pe structura retelei cristaline, pe tipul si distributia gruparilor OH de pe suprafata si legat de acestea pe proprietatile acido-bazice superficiale [75-80].
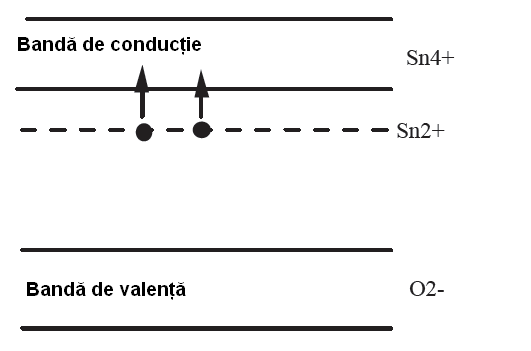
Primul studiu sistematic al naturii si distributiei gruparilor hidroxilice pe suprafata aluminei catalitic active se datoresc lui Peri [75-77] care a elaborat un model statistic al suprafetei aluminei stabilind cinci categorii de oxidrili superficiali dupa numarul de oxigeni adiacenti (0,1,2,3,4 atomi de oxigen), pe baza datelor IR efectuate asupra aluminei.
El a prezentat si un mecanism de formare a alumine prin eliminarea treptata a apei de pe o suprafata ideala total hidroxilata. Conform acestui mecanism , indepartarea gruparilor hidroxil are loc fara ca ordinea locala in reteaua reziduala a oxidului sa fie perturbata, adica fara sa se formeze defecte, pana la indepartarea unei cantitati de 67% din stratul original. Peste aceasta limita , se creeaza defecte in reteauade suprafata, respectiv aparitia a cinci tipuri de grupari hidroxilice izolate (schema 1) care se deosebesc prin configuratia vecinatatii lor imediate.
Schema 1: Schema centrilor acizi si bazici ai -Al2O3
A = grupari OH inconjurate de patru ioni O2-.
D = grupari OH inconjurate de trei ioni O2-.
B = grupari OH inconjurate de doi ioni O2-.
E = grupari OH inconjurate de un singur ion O2-.
C = grupari OH izolate.
Cand deshidratarea este completa , apare o migrare a atomilor pe suprafata care tinde sa minimaliyeye defectele, ceeea ce face ca in final sa predomine trei tipuri de ioni hidroxil.
Cele cinci tipuri de ioni hidroxil difera prin densitatea locala de sarcina, tipul A fiind cel mai negativ (4 ioni de oxigen in vecinatate), tipul C fiind cel mai pozitiv (4 vacante in vecinatate), iar tipul B aproximativ neutru.
S-a considerat ca cele cinci benzi de adsorbtie in IR care apar in domeniul 3700-3800 cm-1 pot fi atribuite celor cinci tipuri de centri, admitand ca frecventa benzilor descresc odata cu micsorarea densitatii electronice respective, in ordinea ADBEC. Rezulta ca centrii A vor fi cei mai bazici, iar centrii C cei mai acizi.
Pe de alta parte, la anumite acoperiri ale suprafetei, alaturi de ioni OH izolati, pot exista si ioni OH invecinati intre care se stabilesc legaturi de hidrogen. La aceste legaturi participa un singur proton, celalalt proton dand nastere in spectrul IR la o banda corespunzatoare hidroxidului izolat, dar cu o frecventa mai joasa. Totodata fiecare ion OH angajat intr-o punte de hidrogen poate fi inconjurat cu 0-3 ioni de oxigen, conducand la aparitia a patru configuratii, fapt ce se va reflecta in frecventa benzilor care creste in functie de numarul de ioni oxigen adiacenti.
Fiecare ion de OH de acest tip va prezenta o reactivitate diferita datorita configuratiei din imediata vecinatate.
Copyright © 2025 - Toate drepturile rezervate