|







 |  |
|
| |
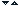 | Alimentatie | Asistenta sociala | Frumusete | Medicina | Medicina veterinara | Retete |
Metabolismul aminoacizilor
Toate celulele contin proteine si nici un act vital nu are loc fara participarea lor. Proteinele pot avea diferite functii cum sunt cea structurala, sau catalitica, sau de transport s.a., dar orice functie presupune uzura in procese cum sunt descuamarea pielii si mucoaselor, degradarea si eliminarea secretiilor proteice. Proteinele se degradeaza pana la aminoacizi pentru a fi sinteti-zate ele insele sau altele. In medie, timpul de injumatatire, T1/2, al proteinelor umane este de circa 80 zile. Intensitatea activitatii metabolice a tesuturilor determina marimea T1/2. Pentru ficat T1/2 = 7 - 10 zile, insa pentru os el este mult mai lung. La organismele tinere sau cele adulte in anumite situatii (de exemplu gestatia) are loc formarea de noi tesuturi ceea ce accelereaza sinteza proteica. Pe de alta parte are loc si trans-formarea ireversibila a proteinelor in produsi ne-proteici.
Pentru aceste nevoi de innoire, dezvoltare si completare, organismul face apel la masa metabolica de aminoacizi. Aceasta contine ami-noacizii liberi din sange (5-8 mg azot%) si din alte lichide extracelulare. Ei provin din dislo-carea tisulara, digestia proteinelor alimentare sau sinteza de novo din compusi neproteici. Organismul uman nu este capabil sa sintetizeze toti cei 20 aminoacizi. 8 dintre acestia sunt aminoacizi esentiali care nu pot fi sintetizati si 2 aminoacizi semiesentiali a caror viteza de sinteza in perioada de crestere este mica si trebuie procurati din surse exogene. Restul de 10 amino-acizi sunt neesentiali (tab.1). Din punct de vede-re nutritiv termenii esential si neesential sunt corecti, adica aminoacizii esentiali nu pot fi obtinuti de organism decat pe cale nutritiva ceea ce sugereaza importanta lor. Din punct de vedere biologic lucrurile par a sta invers, aminoacizii esentiali nu sunt atat de importanti de vreme ce organismul a evoluat spre incapacitatea de a-i sintetiza.
Principalele implicatii medicale legate de aminoacizii neesentiali sunt legate de lipsa sau insuficienta lor in dieta umana. Intrucat anumite cereale au grauntele sarace in triptofan si lizina, in regiunile in care dieta se bazeaza pe aceste plante ca surse de proteine, dieta din care lipseste laptele, pestele, carnea in general, se intalnesc grave deficiente de nutritie. Kwashiorkorul (malnutritia proteocalorica) si marasmul sunt
Tabelul 1. Aminoacizi esentiali, semiesentiali si
neesentiali(*aminoacid semiesential).
|
Esentiali si semiesentiali |
Neesentiali |
|
Arginina* Histidina* Fenilalanina Izoleucina Leucina Lizina Metionina Treonina Triptofan Valina |
Alanina Asparagina Aspartat Cisteina Glicina Glutamat Glutamina Prolina Serina Tirozina |
boli endemice ale Africii de Vest. Kwashiorkorul apare la copilul intarcat pe o dieta bazata pe amidon si saraca in proteine. In marasm sunt deficitare atat caloriile cat si aminoacizii.
Pentru utilizarea aminoacizilor organis-mul nu face discriminare intre aminoacizi endo- si cei exogeni (captati prin absorbtie intestinala). O molecula de aminoacid trece de mai multe ori prin masa metabolica pana cand va iesi din circuit printr-o transformare ireversibila, care poate fi:
dezaminare cu formare de NH3 care este transformat in uree (ureogeneza),
decarboxilare cu formare de amine biogene,
degradarea catenei de carbon urmata de glu-coformare si/sau cetoformare,
transformare in compusi neproteici.
Mentinerea concentratiei constante a amino-acizilor circulanti in sange intre mese depinde de balanta dintre eliberarea lor din stocurile endo-gene si utilizarea lor in diverse tesuturi. Muscu-latura genereaza mai mult de 50% din pool-ul total de aminoacizi in timp ce ficatul este organul in care azotul aminic din aminoacizi este eliberat sub forma de uree. Astfel, muschiul si ficatul joaca un rol important in turnoverul de aminoacizi. Mai mult de 50% din aminonitrogen (azot aminic) este transportat de la muschi la ficat si alte tesuturi de alanina (vezi si ciclul Felig-Malette) si de glutamina. In schimb muschiul capteaza din circu-latia sanguina mici cantitati de serina, cisteina si glutamat. Ficatul si intestinul(tesutul splahnic) capteaza intens alanina si glutamina de la diferite tesuturi extrahepatice. In intestin glutamina pune in libertate NH3. Rinichiul este principala sursa care elibereaza serina, acest organ captand din circulatie glutamina, prolina si glicina. Creierul capteaza in special valina iar capacitatea creierului de sobolan de a oxida aminoacizii cu catena rami-ficata (Leu, Ile, Val) este de 4 ori mai mare decat cea a muschiului si ficatului. In figura 1 apare schematizat schimbul de aminoacizi de la un organ la altul in perioada post-prandiala (post-absorb-tiva) a aminoacizilor.
Zilnic 5-7 g azot proteic se transforma in compusi neproteici si 2-3 g N se elimina din organism. Aceasta cantitate trebuie recuperata pe cale alimentara. Un aport insuficient determina un bilant de azot negativ DN = Ning - Nelim < 0. Aceiasi situatie apare in cazul bolilor degenera-tive, in infectii, hemoragii. La un organism adult sanatos care consuma hrana mixta, DN = 0 (bilant de azot nul). La copii, la care procesele anaboli-zante precumpanesc procesele catabolizante, bi-lantul de azot este pozitiv. Aceiasi situatie o intalnim la convalescentii cu reparatii tisulare sau la femeile gravide.
Figura 1. Schimbul intre organe a aminoacizilor absorbiti digestiv. Se remarca drumul Ala si Gln (sa-getile ingrosate).
Necesarul de proteine alimentare scade cu varsta. Sugarului pana la 3 luni ii sunt necesare zilnic 2,3 g proteine/kgc, iar un adult necesita minimum 0,6 g/kgc. Aceste valori se refera la o proteina de referinta, cu valoare biologica maxima. Alimen-tatia obisnuita a adultului trebuie sa contina 1,2 g proteine/kgc, 1/3 cel putin fiind de origine animala. Cu cat compozitia proteinelor este mai apropiata de cea a proteinelor tisulare cu atat valoarea biologica a lor este mai mare. Astfel, la un adult se poate realiza DN = 0 cu 20 g proteine din oua sau 27 g proteine din lapte, carne sau 42 g proteine din grau (gluten).
1.1. Caile generale de degradare a ami-noacizilor
Spre deosebire de glucide sau lipide, in organism nu exista rezervoare de proteine. Aminoacizii in exces, care nu sunt incorporati in proteine sunt metabolizati sau inglobati in masa metabolica. Conversia aminonitrogenului/NH3 in uree se face cu o viteza apreciabila prin parcurgerea etapelor: dezaminarea oxidativa, trans-dezaminarea, transportul NH3 si ciclul ureogenetic.
Aceasta transformare catalizata de gluta-mat dehidrogenaza (GDH) prezenta in mi-tocondrii este singura care determina dezamina-rea rapida a unui aminoacid, a Glu. Ea are doua etape. Prima este o reactie de dehidrogenare cata-lizata de GDH, NAD(P)+-dependenta, din care rezulta iminoacidul glutamic:
HOOC-CH2-CH2-CH(COOH)-NH2 + NAD(P)+ HOOC-CH2-CH2-CH(COOH)=NH + NAD(P)H++H+
GDH este inhibata de GTP si activata de ADP in vitro ceea ce sugereaza
actiunea acestor nucleotide in vivo
unde enzima functioneaza aproape de echilibru (DG 0). A doua
reactie este hidroliza a-iminoacidului
rezultand acidul a-cetoglutaric (aCG):
HOOC-CH2-CH2-CH(COOH)=NH + H2O HOOC-CH2-CH2-CH(COOH)=O + NH3
Modularea activitatii GDH de ADP si GTP se face in functie de necesitati. Daca un hepatocit necesita intensificarea ciclului citric, dezaminarea oxidativa este intensificata rezultand aCG care este un intermediar al ciclului. Prin urmare, dezaminarea oxidativa a Glu este o reactie anaplerotica. Pe de alta parte, daca, datorita activitatii prea intense a ciclului citric, se acumuleaza GTP, dezaminarea oxidativa a Glu este inhibata.
Datorita etapei 2 secventa celor doua reactii are un bilant energetic defavorabil (DG 30 kJ.mol-1) ceea ce d.p.d.v. fiziologic este impor-tant. Se mentine astfel concentratia NH3 rezultat, substanta toxica pentru sistemul nervos central (SNC), la o concentratie mica.
In afara de GDH exista in ficat si rinichi doua enzime nespecifice, L-aminoacid oxidaza si D-aminoacid oxidaza, cu activitate mica. Reactiile enzimatice catalizate de aceste doua enzime de-curg in doua etape:
a-Aminoacid + FAD + H2O a-Cetoacid + NH3 + FADH2
FADH2 + O2 FAD + H2O2
Apa oxigenata este rapid degradata de catalaza. Existenta D-aminoacid oxidazei este intrucatva enigmatica deoarece D-aminoacizii apar doar in peretele celulelor bacteriene.
Dezaminarea neoxidativa are ca substraturi putini aminoacizi (Ser si His). Reactiile implicate vor fi tratate mai jos.
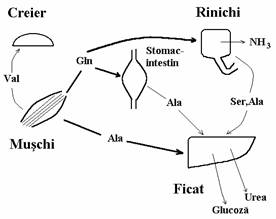
Cuplarea transaminarii catalizata de transaminaze, enzime active in ficat, cord, muschi, cu dezaminarea oxidativa prin interme-diul GDH, asigura dezaminarea rapida a oricarui aminoacid cu exceptia Lys, Thr si Pro (fig. 2).
Figura 2. Trans-dezaminarea, cale de eliminare a aminonitrogenului sub forma de amoniac care este convertit in uree in ciclul ureogenetic
Cum am aratat in volumul I, transami-nazele transfera gruparea -NH2 de la a-amino-acid la a-cetoacid rezultand alt aminoacid si alt cetoacid. Multe transaminaze au a-cetogluta-ratul aCG) drept substrat accep-tor de grupare amino. Acesta se transforma in Glu. Cele mai importante transaminaze din aceasta categorie sunt GOT (glutamic-oxalacetic-transaminaza) si GPT (glutamic-piruvic-transaminaza). Aceste enzime se mai numesc AST (aspartat amino-transferaza) respectiv ALT (alanin aminotrans-feraza). Energia libera standard de reactie, DG 0, ceea ce arata ca transaminarile sunt reversibile. Mai sunt si alte transaminaze ale caror substraturi acceptoare de grupare -NH2 sunt OA sau Pyr. Aminoacizii rezultati sunt Asp respectiv Ala. Prin transaminare se sintetizeaza in organism aminoacizi neesentiali pe seama aminoacizilor esentiali. In concluzie, privind figura 2 putem spune ca daca aminoacizii captati din alimente nu sunt utilizati pentru sinteza de noi aminoacizi, proteine sau alti compusi azotati, Glu este "canalul" prin care gruparea amino este adusa intr-o stare excretabila, prin dezaminare oxidativa si apoi ureogeneza.
1.2. Metabolismul amoniacului
Azotul gruparilor α-NH2(aminonitroge-nul) din structura aminoacizilor se elimina cel mai adesea sub forma de amoniac, in cursul degradarii acestora. Formarea si conversia amo-niacului in structuri netoxice constituie metabo-lismul acestui compus.
1.2.1. Detoxifierea extrahepatica
si transportul NH3
NH3 este un toxic puternic, in special pentru SNC. La pH = 7,4 circa 99% din molecu-lele de NH3 sunt sub forma de ioni NH4+. Acest ion nu poate strabate membrana mitocondriala datorita sarcinii electrice care creeaza asa-zisele "forte imagine" care se opun traversarii membra-nei. Totusi, cei 1% de NH3 penetreaza cu usurinta membrana iar in mitocondrie transforma aCG in Glu, conform reactiei:
aCG + NH3 + NAD(P)H + H+ Glu + H2O + NAD(P)+
Reactia este catalizata tot de enzima GDH. Scaderea concentratiei aCG micsoreaza viteza ciclului acidului citric (CK) si deci a oxidarii glucozei, sursa unica de energie a neuronilor. Rezultatul acestei 'descarcarii' a CK este spolie-rea energetica a neuronilor cu consecinte neuro-logice nefaste, ceea ce explica toxicitatea NH3 la acest nivel. Daca in celulele creierului sau ale altor tesuturi extrahepatice apare NH3 in canti-tati mari el este incorporat rapid in Glu prin ami-narea aCG si apoi este incorporat in Glu-NH2 prin reactiile:
1. HOOC-CH2-CH2-CH(COOH)-NH2 + ATP H2O3POCO-CH2-CH2-CH(COOH)-NH2 + H2O
g-glutamil fosfat
2.H2O3POCO-CH2-CH2-CH(COOH)-NH2 + NH3 H2N- CO-CH2-CH2-CH(COOH)-NH2 +H3PO4
Ambele reactii sunt catalizate de glutamil sinte-taza. Sarcina neta a Glu este -1 la pH-ul fizio-logic, ceea ce impiedica difuzia acestuia prin membrane. In schimb glutamina, a carei sarcina neta este nula, strabate usor membranele difu-zand in sange. Ea este o importanta forma de transport a aminonitrogenului (fig. 1). Eficienta ridicata se datoreaza si continutului ridicat in azot al glutaminei. Ea depaseste de cateva ori concen-tratia plasmatica a celorlalti aminoacizi liberi. Glutamina ajunge pe cale sanguina la ficat unde sub actiunea glutaminazei elibereaza NH3, con-form reactiei:
H2N- CO-CH2-CH2-CH(COOH)-NH2
+ H2O HOOC-CH2-CH2-CH(COOH)-NH2 + NH3
Enzima este prezenta in mitocondriile hepatice. Amoniacul va lua calea ciclului ureogenetic alaturi de el fiind detoxifiat si NH3 rezultat prin dezaminarea Glu.
In musculatura scheletica, in conditii de efort intens, exista o mare productie de NH3 din ami-noacizi si de piruvat rezultat din glicoliza. Fiecare din acesti produsi trebuie sa ajunga la ficat, acest tranzit fiind rezolvat la mamifere printr-un singur ciclu: ciclul glucoza-alanina (Felig-Malette). In muschi NH3 este detoxifiat prin transformare in Glu care prin transaminare cu Pyr formeaza Ala. Aceasta este molecula care transporta azotul la ficat unde transaminarea in sens invers reface Pyr si Glu.
1.2.2. Ciclul ureogenetic (ciclul Krebs-Henseleit)
Am vazut ca formele de detoxifiere extrahepatica si de transport a NH3 pana la ficat sunt aminoacizii Glu, Glu-NH2 si Ala. In ficatul animalelor ureotelice este sintetizata forma finala de detoxifiere a NH3 , forma de excretabi-la care este ureea.
Scurt istoric. Acest ciclu a fost desco-perit de Hans Krebs si un student in medicina, Kurt Henseleit, in 1932. Cinci ani mai tarziu Krebs a postulat ciclul citric. Cei doi au observat ca viteza cu care foite de ficat, suspendate intr-un mediu aerat, transforma amoniacul in uree, creste prin adaugarea ornitinei, citrulinei sau argininei. Structurile celor 3 aminoacizi au sugerat o secventa de trans-formari in care ornitina este precursor al citrulinei iar aceasta precede argini-na. Se stia mai demult ca prin hidroliza Arg se formeaza ornitina si uree. Din aceste date Krebs a dedus ca are loc un proces ciclic. Mai tarziu el a vazut ca ornitina joaca un rol asemanator cu cel al oxalacetatului in ciclul citric.
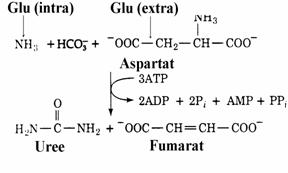
Hepatocitele incorporeaza intr-o molecula de uree doi atomi de azot dintre care unul provine din amoniacul din mitocondrie iar celalalt pro-vine dintr-o molecula de aspartat din citosol. Indirect cei doi atomi de azot provin din gluta-matul intramitocondrial care se dezamineaza si glutamatul extramitocondrial care, prin transami-nare catalizata de GOT formeaza aspartatul (fig. 3).
Figura 3 Reactia globala a ciclului ureogenetic.
In figura 4 este prezentat mecanismul chimic al ciclului ureogenetic.
1. Sinteza carbamoilfosfatului. NH3 re-zultat din dezaminarea oxidativa a Glu este incor-porat in carbamoilfosfat, prin reactia cu CO2 rezultat din respiratia celulara. Reactia este con-sumatoare de energie metabolica si este catalizata de carbamoil fosfat sintetaza (I) mitocondriala care se distinge de cea citosolica necesara in bio-sinteza nucleotidelor pirimidinice (carbamoil fos-fat sintetaza II).
Carbamoilfosfat sintetaza I este o enzima premergatoare ciclului ureogenetic, carbamoil-fosfatul fiind un compus macroergic care poate fi privit ca un donor de grupare carbamoil care introduce aceasta grupare in ciclu. Carbamoil-fosfat sintetaza I este o enzima reglatoare stimu-lata de N-acetil-Glu care modifica afinitatea enzi-mei pentru ATP (vezi reglarea ciclului).
2. Sinteza citrulinei. Se face prin transfe-rul gruparii carbamoil pe ornitina. Enzima, orni-tin transcarbamoilaza prezenta in mitocondrie are drept substrat un aminoacid "nestandard".
3. Sinteza acidului argininosuccinic. Ci-trulina paraseste mitocondria si trece in citosolul hepatocitului. Al doilea atom de azot este preluat de la Asp printr-o reactie de condensare cataliza-ta de argininosuccinat sintetaza, enzima citoso-lara. Reactia este insotita de rearanjari atomice ceea ce necesita energie libera. De aceea ea este cuplata cu hidroliza PPi. Asa se explica modul in care pool-ul glutamic intramitocondrial introduce in ciclul ureogenetic un atom de azot iar pool-ul glutamic citosolic introduce celalalt atom de N ai moleculei de uree.
4. Clivajul acidului argininosuccinic. Se produce sub actiunea argininosuccinat liazei, cu
formare de fumarat care reintra in mitocondrie pentru a incarca ciclul lui Krebs.
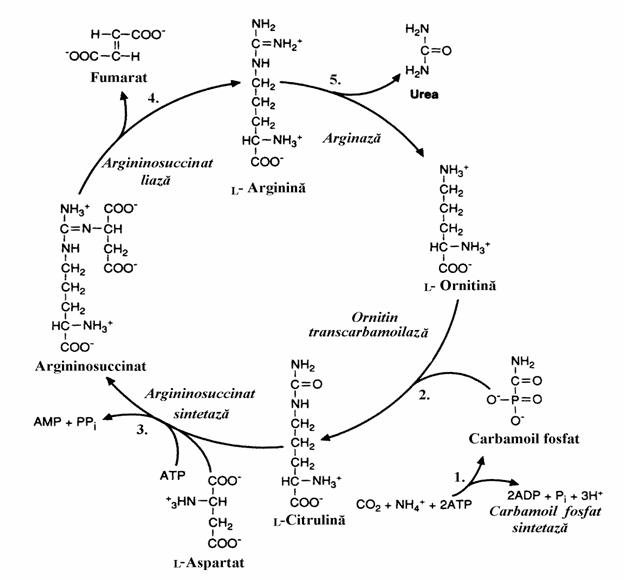
5. Hidroliza argininei. Cu
aceasta reactie
catalizata de arginaza se inchide ciclul cu regenerarea ornitinei si eliberarea ureei. Datorita permeabilitatii membranelor pentru uree aceasta difuzeaza in sange si, de aici, prin filtrul glomerular ajunge in urina. La randul ei, ornitina patrunde in mitocondrie reluand ciclul ureo-genetic. Fumaratul realizeaza o legatura intre cele 2 cicluri Krebs. Astfel, dupa ce reintra in mito-condrie, el se integreaza in ciclul acidului citric conducand la oxalacetat care, dupa extrudere (ca citrat sau malat) poate reface Asp prin transami-nare. Integrarea dezaminarii oxidative cu trans-aminarea si cele 2 cicluri krebsiene, pentru ex-cretia azotului ureic apare in schema din figura 5.
Figura 4. Reactiile ciclului ureogenetic. Exista o asemanare intre acesta si ciclul citric in ce priveste rolul "catalizator" al intermediarilor. Ele se deose-besc prin faptul ca primul serveste la o sinteza (a ureei) iar celalalt la o degradare(a acetilului).
Figura 5. Integrarea transdezaminarii si a celor doua cicluri krebsiene. Ciclul ureogenetic si ciclul acidului citric sunt cuplate prin formarea si scindarea argi-ninosuccinatului. Fumaraza (1) si malat dehidroge-naza (2) sunt enzime ale ciclului citric. Oxalacetatul este extras din ciclul lui Krebs pentru a forma aspar-tat sub actiunea GOT (3). ATP este hidrolizat in reactiile catalizate de carbamoilfosfat sintetaza I (5) si argininosuccinat sintetaza (6). Acest ATP este regenerat prin fosforilare oxidativa din NAD(P)H-ul produs in reactiile catalizate de glutamat dehidroge-naza (4) si malat dehidrogenaza (2).
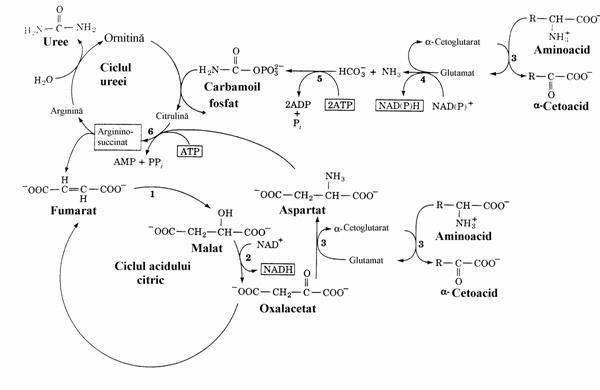
Reactiile cu formare de aCG si Fum au caracter anaplerotic. Se vede ca Glu este un "canal" pentru N-ul aminic al aminoacizilor. El este un "dispecer" metabolic: poate introduce azotul di-rect in ciclul ureogenetic ca NH3 sau prin inter-mediul Asp, ceea ce implica un cuplaj cu CK.
Cum am mai aratat activitatea GDH este reglata alosteric de nucleotidele libere: ADP-ul o activeaza si GTP-ul o inhiba. Prin urmare, o scadere a incarcarii energetice a celulei acce-lereaza dezaminarea Glu dar si ciclul citric, ceea ce mareste fluxul azotului pe cele 2 cai de incorporare in uree (muschiul in efort). Aceasta este o reglare indirecta a ciclului ureogenetic. Reglarea directa este realizata de N-acetil-glutamatul care activeaza prima etapa a ciclului prin efect alosteric asupra carbamoil fosfat sinte-tazei. Acest metabolit este sintetizat din Glu si CH3COSCoA sub actiunea N-acetilglutamat sintazei si este hidrolizat de o hidrolaza specifi-ca. Viteza de sinteza a ureei in ficat este determi-nata de concentratia acestui efector alosteric.
1.2.2.3. Bilantul energetic al ciclului ureogenetic
Reactia globala a ciclului ureogenetic este urmatoarea:
2 NH3 + CO2 + 3ATP + H2O uree + 2(ADP+Pi) + AMP + PPi
In reactia de mai sus se vede ca sinteza unui mol de uree necesita 4 moli de legaturi macroergice: 2 ale fosfatului g, una a fosfatului b din ATP si una a PPi. Procesul este deci mare consumator de energie metabolica. Este intere-sant de observat ca scaderea incarcarii energe-tice a celulei intensifica un proces endergonic. Contradictia este aparenta. Privilegiul de a excreta uree in loc de NH3 le costa pe animalele ureotelice doar 15% din energia aminoacizilor care si-au eliberat aminonitrogenul. Catena de carbon a acestora furnizeaza suficienta energie prin degradare.
In aceasta ordine de idei, animale amo-notelice sunt pestii si majoritatea speciilor acva-tice iar animale uricotelice sunt pasarile si serpii. Aceste diferente in mecanismul de eliminare a reziduului de azot se bazeaza pe specificul anatomo-fiziologic si pe mediul de viata al aces-tor animale. In pesti glutamina este hidrolizata in bronhii, NH3 rezultat fiind diluat in marele volum de apa vehiculat de bronhii. De aceea speciile acvatice nu au nevoie de un sistem urinar. In scara filogenetica unele specii s-au acomodat vietii terestre. Acestea si-au dezvoltat alte cai de excretie a azotului aminic. Ele sunt prevazute cu rinichi si un rezervor, vezica urinara. Intrucat NH3 difuzeaza liber prin membrane excretia in urina poate fi insotita si de resorbtia acestuia in sange. Excretia masiva a ionului NH4+ ar necesita pentru prevenirea crearii unui gradient electric, excretia unui numar echivalent de anioni: Cl- sau Pi. Pentru a evita aceasta eliminare, animalele terestre excreta ureea, compus neutru, netoxic, foarte solubil. Aceasta cale, am vazut, solicita consum de energie metabolica.
In ce priveste animalele uricotelice, acestea in cursul evolutiei au invatat sa excrete aminonitrogenul intr-o forma care sa fie cat mai usoara si sa ocupe un volum cat mai mic. Pasarile excreta acidul uric, substanta relativ insolubila, intr-o stare de pasta cu mic continut de apa. Costul energetic al sintezei acidului uric este mai ridicat decat al ureei, in primul fiind condensati 4 atomi de azot.
1.2.2.4. Tulburari ale echipamentului enzimatic al ciclului ureogenetic
Intrucat nu se cunosc cai alternative pentru sinteza ureei, blocarea completa a oricarei etape din ciclul ureogenetic este incompatibila cu viata. Decesul ar surveni datorita intoxicarii cu NH3. Sunt cunoscute deficiente ale tuturor enzimelor din echipamentul enzimatic al ciclului ureoge-netic. Intreruperea acestui ciclu in diferite puncte ale sale afecteaza in mod specific metabolismul azotului deoarece precursorii corespunzatori difuzeaza din hepatocit in sange si trec in urina determinand diferite simptome. Bolile determi-nate de aceste deficiente sunt grave avand mari incidente. Simptomele sunt retardarea mintala, apoplexie, coma, moarte timpurie.
1. Deficienta de carbamoilfosfat sintetaza. Cum am vazut, N-acetil glutamatul este un activator al carbamoil fosfat sintetazei. La un nou nascut avand deficitara aceasta enzima adminis-trarea carbamoil-glutamatului, un analog al N-acetilglutamatului, a condus la normalizarea metabolismului azotat. In aceasta deficienta (0-50% din activitatea normala a enzimei) principa-lul simptom primar este hiperamonemia de tip I. Ca terapie se aplica dieta saraca in proteine. Totusi la acesti copii apare retardarea mintala ca o consecinta indirecta a perioadelor de hiperamo-nemie necontrolata.
2. Deficienta de ornitin transcarbamoilaza. Este cea mai frecventa boala data de deficienta unei enzime ureogenetice. Principalul simptom paraclinic este hiperamonemia de tip II care este prevenita la fel ca in cazul precedent. Gena responsabila de biosinteza acestei enzime este localizata pe cromozomul X. Deci, barbatii sunt mai puternic afectati decat femeile care, daca sunt heterozigote, deseori sunt asimptomatice. Bolnavele manifesta aversiune fata de alimentele bogate in proteine.
3. Deficienta argininosuccinat sintetazei. Incapacitatea metabolizarii citrulinei determina difuzia acesteia in sange si apoi excretia in urina pana la valori care arata ca majoritatea azotului excretat este sub forma de citrulina (citrulinote-lie).
4. Deficienta argininosuccinat liazei. In acest caz sunt excretate in urina mari cantitati de argi-ninosuccinat.
5. Deficienta arginazei. In acest caz se acumuleaza si este excretata arginina. Totusi sunt excretate si cantitati de uree datorita exis-tentei unei arginazei renale cu activitate relativ redusa.
In conditiile pierderii severe a functiunilor hepa-tice (ciroza) capacitatea ureogenetica a ficatului poate fi depasita de productia de NH3 ceea ce conduce la aparitia comei hepatice datorita cres-terii amonemiei din circulatia sistemica pana la nivele toxice.
In general decarboxilarea unui aminoacid proteinogen sau neproteinogen monocarboxilic produce o amina biogena (ptomaina) care are importante proprietati biologice. Cateva exemple puse intr-o reprezentare schematica apar in figura 6.
Figura 6. Decarboxilarea Glu si His.
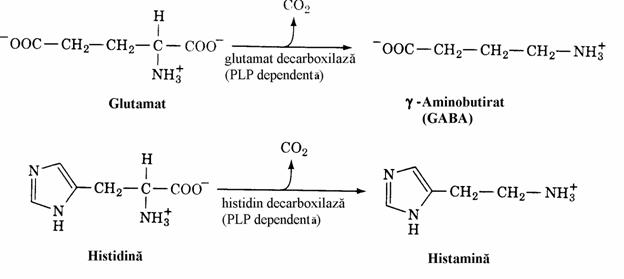
Decarboxilarea acidului glutamic da nas-tere acidului g-aminobutiric (GABA) iar din his-tidina, prin decarboxilare, rezulta histamina.
Glu si Gln sunt principalele surse de sinteza a GABA in creier, cei doi aminoacizi continand cca 60% din aminonitrogenul cerebral. Mentinerea concentratiei acestuia se realizeaza pe calea asa-zisului "sunt GABA", circuit meta-bolic care produce o molecula de Glu pentru fiecare molecula de GABA folosita in neuro-transmisie. Adica, dupa sinteza din Glu/Gln a GABA in terminatia nervoasa a unui neuron presinaptic, neurotransmitatorul, care si-a exer-citat functia inhibitorie in sinapsa, este captat de o celula gliala vecina in care prin transaminare da nastere seminaldehidei succinice. Acceptorul gruparii amino a acestei reactii este a-cetoglu-taratul care se transforma in Glu si apoi in Gln. Semialdehida se oxideaza la acid succinic, intermediar krebsian al celulei gliale. GABA este un neurotransmitator inhibitor care actioneaza asupra neuronilor post-sinaptici printr-un meca-nism ce implica un canal pentru ionii Cl-, parte componenta a receptorului GABA-ergic de tip A (GABAA). Receptorul GABAA joaca un impor-tant rol in inhibitie in SNC necesara functiilor de coordonare si excitabilitate controlata. Actiunea inhibitorie a GABA se bazeaza pe deschiderea canalului de Cl- si patrunderea acestor ioni, contra gradientului electric transmembranar, in neuronul post-sinaptic. Hiperpolarizarea mem-branei datorata acestui curent (de anioni) post-sinaptic are loc pana cand cele doua potentiale, chimic si electric, devin egale. Ea micsoreaza excitabilitatea neuronului post-sinaptic adica il inhiba. Din punct de vedere medical deficitul de GABA este uneori asociat cu crizele de epilepsie. Amina biogena este utilizata in tratamentul epilepsiei si hipertensiunii. Evident, perturbarile functionalitatii receptorilor GABA-ergici se asociaza cu diferite boli ca de exemplu boala Parkinson, chorea Huntington,s.a. Glicina, neu-rotransmitator inhibitor, actioneaza printr-un mecanism similar. Histamina, produsa in special in celulele mastocite, este un puternic vasodila-tator dar si vasoconstrictor. Ea este responsabila de reactiile alergice atunci cand este eliberata in cantitati excesive. Aceste reactii pot fi declansate de medicamente cum ar fi penicilinele ceea ce impune testarea pre-terapeutica a bolnavilor cu aceste medicamente. Cea mai dramatica reactie alergica imediata (2-30 minute dupa administra-rea agentului alergen) este anafilaxia. Simpto-mele in anafilaxie sunt anxietate, senzatie de fierbinteala, urmate de acuze de presiune sub-sternala, dificultati de respiratie, prabusire a tensiunii arteriale si anoxie. Moartea poate sur-veni in cateva minute. Edemul obstructiv a caii respiratorii superioare, laringospasmul si bronho-spasmul sunt principalele cauze ale mortii. Meca-nistic, anafilaxia are o importanta componenta imunoumorala implicand anticorpii IgE. Acestia sunt produsi ca raspuns la prezenta alergo-genului. Ei au o mare afinitate pentru receptorii de suprafata ai celulelor mastocite (fig. 7).
Figura 7. Degranularea mastocitara in reactia anafilactica. Legarea incrucisata a anticorpilor IgE fixati pe suprafata celulelor mastocite declanseaza eliberarea continutului granulelor mastocitare. SRS-A = substante lent-reactive ale anafilaxiei.
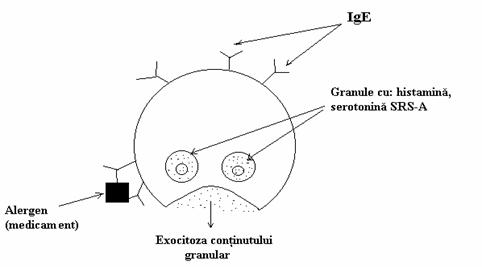
Legarea incrucisata (cross-linking) a moleculelor de IgE prin intermediul moleculelor alegenului produce eliberarea masiva de hista-mina, prostaglandine, serotonina, peptido-leuco-triene (SRS-A). Acesti mediatori actioneaza asupra musculaturii netede si tesutului vascular cauzand simptomele socului anafilactic. Inciden-ta anafilaxiei cauzate de peniciline este de cca 0,04% iar tratamentul consta in administrarea adrenalinei si a perfuzabilelor izotonice pentru refacerea volemiei precum si asigurarea functio-narii cailor respiratorii.
Efectele histaminei asupra tubului digestiv depind de tipul receptorilor histami-nergici (H1 sau H2). La nivelul celulelor parietale stomacale asocierea aminei biogene la receptorii H2 determina cresterea secretiei gastrice pe cand asocierea la receptorii H1 o reduc. La nivelul aparatului circulator histamina de origine mastocitara exercita efecte de tip stimulator inotrop si cronotrop-pozitive. Asocierea ei la receptorii H1 determina contractia muschiului neted de la nivelul vaselor mari si dilatarea partiala la nivelul vaselor capilare. Asocierea la receptorii H2 determina vasodilatatie generali-zata. Efectele contracturante de la nivelul musculaturii bronsice si uterine sunt rezultatul asocierii histaminei la receptorii H1, asocierea la cei de tip H2 determinand in aceleasi teritorii miorelaxare. La nivelul structurilor nervoase centrale si periferice asocierea histaminei la receptori H1 are efect excitator iar asocierea la receptori H2 are efect inhibitor. Introducerea H2-
antagonistilor ai histaminei in tratamentul ulce-relor si hiperaciditatii gastrice este una din cele mai importante aplicatii. Un exemplu de antago-nist este cimetidina cunoscuta si sub denumirea comerciala Tagamet . Cimetidina inhiba meta-bolismul hepatic mediat de sistemele enzimatice citocrom P450-dependente, ceea ce ridica pro-blema unor precautii terapeutice. Administrarea concomitenta a cimetidinei cu medicamente ca warfarina (trombostop), benzodiazepine (diaze-pam) s.a. potenteaza actiunile terapeutice ale celor din urma. Ranitidina, un alt H2-antagonist, interfera in mai mica masura cu oxidarile citP450-dependente, deci si cu eliminarea acestor medica-mente. In fine, antidepresivele tri- si tetraciclice sunt antagonisti competitivi ai receptorilor H1- si H2 -histaminergici iar dintre neuroleptice clor-promazina este un H2- antagonist.
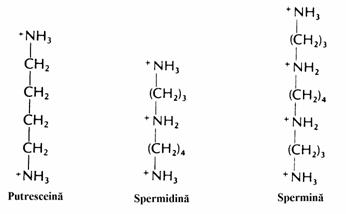
Putresceina este produsul de decarboxi-lare al ornitinei iar cadaverina rezulta prin decarboxilarea Lys. Decarboxilazele care catali-zeaza formarea acestor doua diamine toxice se gasesc in bacteriile florei intestinale. Aceste diamine contribuie la mirosul caracteristic al cadavrelor.Alte reactii de decarboxilare ale unor produsi de metaboli-zare ale aminoacizilor vor fi prezentate mai jos. Acestea au o deosebita importanta biologica deoarece produc compusi bioactivi (serotonina, catecholamine) sau polia-mine ca spermidina si spermina (fig. 8).
Figura 8. Structurile chimice ale putresceinei, spermidinei si sperminei.
Alte reactii de conversie a aminoacizilor in ptomaine:
Arg Agmatina + CO2 Asp b-Ala+ CO2 Cys Cisteamina + CO2
Ser Etanolamina + CO2 Thr Propanolamina+ CO2 Tyr Tiramina + CO2.
In afara reactiilor catalizate de decarboxi-lazele bacteriene, in intestin se produc si alte reactii de fermentatie si putrefactie care produc diferite gaze (CH4, N2, H2S, s.a.) precum si acizi carboxilici (acetic, lactic, butiric).In intestin trip-tofanul sufera o serie de reactii pentru a forma indolul si metilindolul (scatol), substante care contribuie la mirosul fecalelor. Similar, Cys se transforma in alchilmercaptani, metilmercap-tan (CH3SH) si etilmercaptan (CH3CH2SH). Metil-mercaptanul prin hidrogenare produce CH4 si H2S. In intestinul gros se produc cantitati consi-derabile de amoniac care este absorbit in circula-tia portala si, in conditii normale, este rapid con-vertit in uree. In bolile ficatului aceasta capacitate de detoxifiere a NH3 este redusa si amonemia poate atinge nivele toxice. Intoxicatia cu amoniac poate conduce la instalarea comei hepatice.
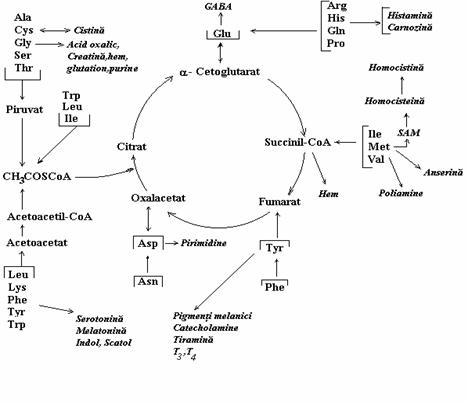
Degradarea aminoacizilor poate conduce la intermediari ai ciclului citric sau precursori ai acestora (fig. 9). Avand in vedere caracterul amfibolic al ciclului citric soarta scheletelor de carbon a aminoacizilor poate fi aceea de sinteza a
glucozei (glicogenului) in cazul aminoacizilor glucoplastici, aceea de sinteza a corpilor cetonici (cetoplastici) si aceea de sinteza a glucozei si corpilor cetonici (gluco- si cetoplastici, micsti), asa cum apare in tabelul 2. Degradarea oxidativa a aminoacizilor furnizeaza 10-15% din energia metabolica la animale.
Totusi aminoacizii nu constituie o sursa majora de energie asa cum am vazut ca sunt glucidele si lipidele.
Tabelul 2. Clasificarea aminoacizilor in functie de soarta scheletelor atomilor de carbon.
|
Glucoplastici |
Cetoplastici |
Micsti |
|
Ala, Arg, Asn, Asp, Cys, Gln, Glu, Gly, His, Met, Pro, Ser, Thr, Val |
Leu, Lys |
Ile, Phe, Trp, Tyr |
Figura 9. Metabolizarea scheletelor aminoacizilor. GABA= acid g-aminobutiric; SAM= S-adenozilmetionina; T3 si T4= tri- si tetraiodotironina
 |
In piruvat (Pyr) se incorporeaza atomii de carbon din 6 aminoacizi (Ala, Cistina, Cys,
Gly, Ser si Thr) (fig. 10), toti fiind stereoizomeri L
Figura 10. Caile de degradare ale Ala, Cistinei ,Cys,Gly,Ser si Thr in piruvat. Enzimele implicate sunt: alanin amino-transferaza (GTP)(1). serin dehidrataza (2), sistemul de clivaj al glicinei (3), serin hidroximetil trans-feraza (4 si 5), cistin reductaza (6).
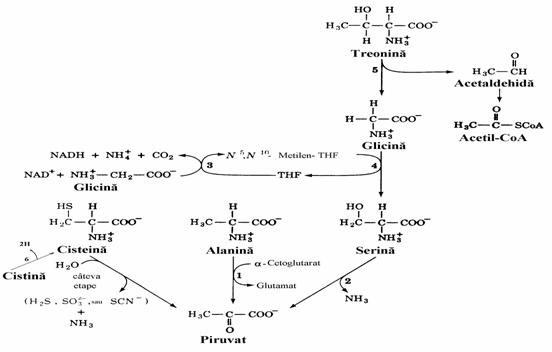
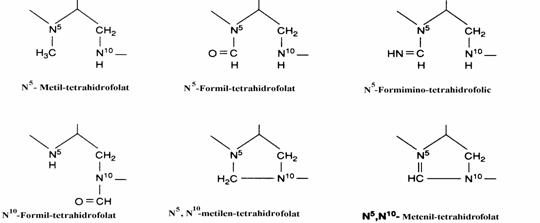
Toti atomii de carbon participa la formarea Pyr in cazul a 5 aminoacizi. Exceptie face Thr care participa doar cu 2 atomi de carbon. Conform figurii Thr ar fi un aminoacid mixt. Totusi, o cale alternativa de catabolizare implica conversia: Thr aminoacetona + CO2 + 2H. Ulterior amino-acetona se transfomra tot in Pyr. In concluzie intrucat conversia Pyr in glucoza apartine gluco-neogenezei, cei 6 aminoacizi sunt glucoplastici. Conversia Ala in Pyr este catalizata de o transaminaza (GPT). Enzima care catalizeaza transformarea Ser in Pyr, serin dehidrataza, este PLP-dependenta. Amoniacul se elimina in final prin hidroliza spontana. Glicina se transforma in serina sub actiunea serin hidroximetil transferaza, alta enzima PLP-dependenta, care utilizeaza drept cofactor N5,N10 - metilen-tetrahidrofolat. Acesta este un donor de unitate C1 necesara aces-tei conversii. La randul ei, glicina se poate des-compune sub actiunea unui sistem de clivaj. Atunci cand actioneaza in sens invers, sistemul de clivaj al glicinei se numeste glicin sintaza.In ce priveste unitatea C1 ea este transferata de acidul tetrahidrofolic (FH4). Atomul de carbon (C1) prezent in structura folica poate fi in diferite stari de oxidatie (fig. 11) exceptand starea +4 (CO2).
Figura 11. Unitatea C1, in diferite stari de oxidatie, transportata de acidul tetrahidrofolic.
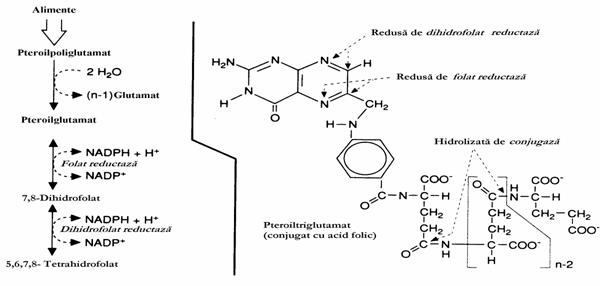
Incapacitatea celulelor umane de a sinte-tiza scheletul pteroil (pteridina-PABA) face ca acesta sa fie dobandit pe cale dietetica sub forma conjugata cu tripeptidul triglutamat, pteroiltri-glutamat, un precursor al FH4 (fig. 12).
Figura 12. Formarea FH4. In mucoasa intestinala sunt indepartate resturi Glu cu exceptia unuia. Pteroilglutamatul este redus cu putere reducatoare pana la FH4. In partea dreapta a figurii sunt precizate structurile din pteroiltriglutamat asupra carora actioneaza diferitele enzime din mucoasa intestinala.
Pteroilglutamatul format in tubul intestinal se reduce progresiv
pana la FH4. Serin hidroximetil transferaza, actionand in
sensul invers reactiei (4) din figura 10, scindeaza Thr in Gly
si acet-aldehida. Aceasta reactie nu necesita FH4
ca acceptor de acetaldehida, aldehida transforman-du-se in CH3COSCoA.
Cisteina poate fi transformata in Pyr pe mai multe cai pe care se
pot elibera compusi ai sulfului (H2S, SO32-
sau SCN-) si amoniac. Prin reducere cisteina se transforma
in cistina. In ce priveste patologia catabolismului acestor
amino-acizi, glicinuria este
caracterizata printr-o excretie excesiva de Gly in urina
si tendinta de formare a calculilor renali pe baza de oxalat ce
calciu (nefrocalcinoza) datorita conversiei Gly acid oxalic. Hiperoxaluria
primara consta din excretia continua si intensa a
acidului oxalic rezultat prin dezaminarea si oxidarea Gly care constituie
conversia aratata mai sus. Consecintele sunt urolitiaza
(calculii vezicii urinare si ai tractului urinar) pe baza de oxalat
de calciu, nefrocalcinoza, infectia recurenta a tractului uri-nar
si moartea inca din copilarie a bolnavului. Daca bolnavii
ating varsta adulta moartea inter-vine datorita insuficientei
renale si/sau hiperten-siunii. In boala innascuta numita hiper-glicine-mie necetonica principalul simptom este retar-darea mintala
datorata acumularii unor mari cantitati de Gly in lichidele
organismului ca rezultat al absentei sistemului de clivaj al Gly.
Dintre bolile metabolice care implica aminoacizi cu sulf prezentam cateva. In cistinu-rii excretia urinara a cistinei este de zeci de ori mai mare decat la sanatosi. Datorita solubilitatii mici a cistinei, la bolnavi acest compus precipita formand calculi in tubulii renali. In cistinoza (boala de stocaj a cistinei) cristalele de cistina sunt depozitate in mai multe tesuturi si organe, in special in sistemul reticuloendotelial. Bolnavii mor de timpuriu datorita insuficientei renale acu-te.
1.3.2.2. Aminoacizi care se transforma in oxalacetat
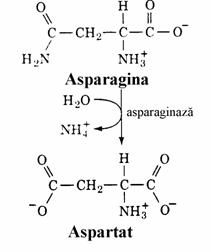
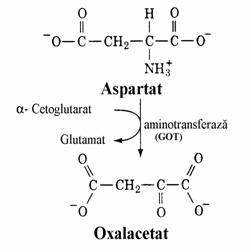
Transaminarea Asp conduce direct la oxalacetat iar asparagina este convertita in Asp sub actiunea asparaginazei (fig. 13).
Figura 13. Conversia aminoacizilor in oxalacetat.
1.3.2.3. Aminoacizi care se transforma in a-cetoglutarat
Arginina, glutamina, glutamatul, prolina si histidina, alaturi de care si aminoacidul "ne-standard", ornitina, se transforma in a-cetogluta-rat (aCG) (fig. 14). Intrucat cetoacidul este intermediar krebsian, acesti aminoacizi participa la anapleroza ciclului citric.
Figura 14. Caile de degradare ale Arg, Glu, Gln, His si Pro in a-cetoglutarat. Enzimele implicate sunt: glutamat dehidrogenaza (1), glutaminaza (2), argina-za (3), ornitin-d-aminotransferaza (4), glutamat semi-aldehida dehidrogenaza (5), prolin oxidaza (6), spontan (7), histidin liaza (8), urocanat hidrataza (9), imidazolon propionaza (10) si glutamat formimino transferaza (11).
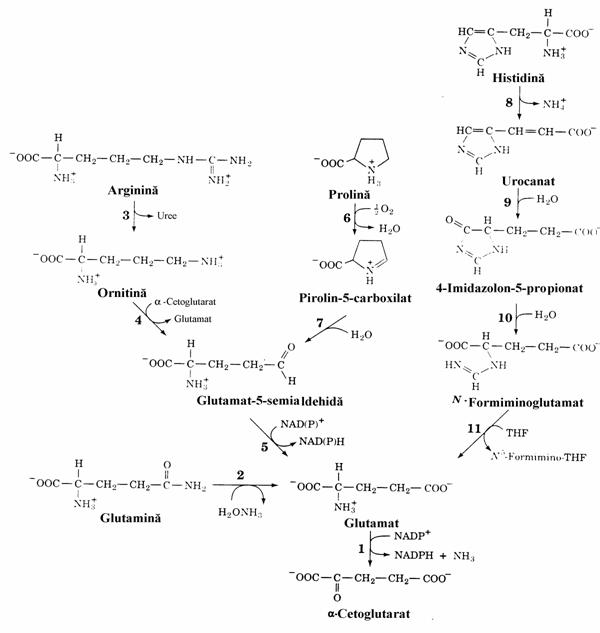
Glutamina se transforma in Glu prin hidroliza sub actiunea glutaminazei. Transformarea Arg ornitina este similara reactiei 5 din ciclul ureogenetic (fig. 4). Conversia histidinei in Glu este mai complicata: ea implica dezaminarea neoxidativa a His (reactia catalizata de enzima 8
din figura), hidratare, spargerea ciclului imida-zolic cu formarea N-formiminoglutamat si transferul gruparii formimino la acidul tetra-hidrofolic pentru a rezulta N5-formimino-tetra-hidrofolat (fig. 11) si Glu. Atat arginina cat si prolina se transforma in glutamat prin interme-
diul glutamat-5-semialdehidei. Dupa cum se vede in figura, conversia in aCG implica Glu ca un intermediar comun.
Din punct de vedere patologic, nu sunt cunoscute defecte metabolice ale caii catabolice glutamina-glutamat. In schimb, se cunosc doua hiperpro-linemii distincte din punct de vedere genetic. In cea de tip I blocajul metabolic este la nivelul prolin oxidazei. Heterozigotii prezinta nivele doar putin crescute ale prolinemiei. In boala de tip II deficitul enzimatic este al glutamat-
semialdehid dehidrogenazei. Heterozigotii acestui deficit nu prezinta hiperprolinemie. Tul-burarea metabolica numita hiperargininemie este cauzata de deficitul de arginaza hepatica, enzima ureogenetica. Histidinemia este o boala inascuta in care mai mult de jumatate din bolnavi prezinta retardare mintala si un defect de vorbire caracteristic. Nivelul His este crescut in sange si urina. Enzima deficitara este histidin liaza motiv pentru care are loc transaminarea neobisnuita a L His, o "diversiune" metabolica ce duce la excre-tia crescuta in urina a imidazol piruvatului. Testul de laborator este o reactie de culoare cu FeCl3 care, daca este pozitiv se poate datora si altei tulburari metabolice, fenilcetonurie (vezi mai jos), in care se excreta cantitati mari de fenil-piruvat tot ca urmare a unei "diversiuni" meta-bolice. Alaturi de imidazol-piruvat se excreta in urina si cantitati patologice de imidazol-lactat, produsul de hidrogenare al celui dintai.
O crestere surprinzatoare a excretiei de His apare in timpul sarcinii. Ea reflecta modificarile func-tiilor renale ale femeii insarcinate.
1.3.2.4. Aminoacizi care produc succi-nil-CoA
Metionina, valina si izoleucina parcurg cai metabolice complexe pentru a produce propionil-CoA (fig. 16 si 19), produs principal al b-oxidarii acizilor cu numar impar de atomi de carbon. Conversia CH3CH2COSCoA in succinil-CoA a fost descrisa in vol. II, totusi din motive ce vizeaza patologia ei, o reluam cu precizarea ca etapa de izomerizare (S) (R) catalizata de metilmalonil-CoA racemaza poate fi definita si ca o reactie de izomerizare D L (fig. 15). Reamintim ca succesiunea de reactii prin care radicalul propionil se transforma in succinil nece-sita biotina si coenzina B12.
Figura 15. Transformarea propionil-CoA in succinil-CoA. Conversia "D L" a metilmalonil-CoA este catalizata de o racemaza.
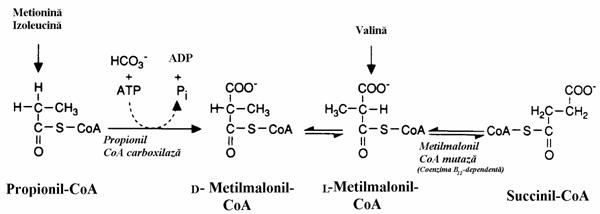
Aceasta reactie este necesara nu numai in catabolizarea aminoacizilor mentionati ci si
in cea a compusilor izoprenoizi si a catenei laterale a colesterolului in cadrul procesului de transformare a acestuia in acizi biliari. Acidul propionic poate fi ingerat pe cale alimentara. Astfel, un tip de branza elvetiana este preparata prin cultivare de bacterii care produc acest acid iar in paine se introduc mici cantitati de acid propionic ca fungicid
Din punct de vedere patologic, deficitul de propionil-CoA carboxilaza produce acide-mia propionica in care concentratia serica a propionatului este mare, ea fiind una din acido-zele metabolice. Totodata, este implicata aici si
incapacitatea de catabolizare leucocitara a acestui acid. Tratamentul se bazeaza pe aplicarea unei diete sarace in proteine. Aciduria metil-maloni-ca, o alta acidoza metabolica, este de doua tipuri: in unul din acesta ameliorarea starii bolnavului depinde de administrarea intramusculara a unor doze masive de vitamina B12. Un caz tipic a fost acela a unei studente eminente care a suferit o deteriorare subita. Pe parcursul unui an capacitatea mintala a scazut dramatic, mersul ei a devenit dezechilibrat iar in urina i-au aparut mari cantitati de acid metilmalonic si homocisteina. Tratamentul cu mari cantitati de vitamina B12 a eliminat tulburarile biochimice si functionale. Totusi, dupa cateva luni au aparut semne de mielopatie. Variatiile parametrilor biochimici in acidoza metilmalonica sunt mari.
Astfel, pH-ul sangelui arterial scade pana aproape de 7,0 iar excretia metilmalonatului creste puternic. Administrarea de vitamina B12 amelioreaza aceste simptome. Cauza acestui tip de acidoza este deficitul de activitate a metil-malonil-CoA mutazei datorita lipsei partii pros-tetice a acesteia, coenzima B12. La baza este un deficit de transferaza care, catalizeaza sinteza coenzimei B12 din vitamina B12 (cobalamina) si ATP: B12 (Co+) + ATP -deoxiadenozilco-balamina + Pi + PPi. Cauza cresterii homocis-teinei in urina studentei prezentata mai sus vizeaza o alta enzima cobalamin-dependenta, deficitul homocistein metiltransferazei. Al doilea tip de acidurie metilmalonica raspunde terapeutic la administrarea parenterala a unor doze mai mici de vitamina B12. Bolnavii care nu raspund la tratamentul cu vitamina B12 pot avea deficitara apoenzima metilmalonil-CoA mutazei. Aceasta forma de acidurie metilmalonica este de obicei letala. Pacientilor care nu raspund la tratamentul cu vitamina B12 li se poate modifica dieta prin reducerea ingestiei de proteine care contin cei 3 aminoacizi producatori de propionil-CoA. Totusi, managementul acestei terapii este dificil deoarece Met,Val si Ile sunt aminoacizi esentiali, absolut necesari cresterii si dezvoltarii.
In figura 17 este prezentata detaliat con-versia Met in succinil-CoA. Reprezentarea schea-matizata a acesteia, cu evidentierea ciclului meti-lului activat, apare in figura 16. Calea metabo-lica debuteaza cu sinteza S-adenozil-metioninei (SAM) in care gruparea metil, foarte reactiva, a cationului sulfonium participa la importante re-actii de metilare cum ar fi sinteza lecitinelor din cefaline sau conversia noradrenalinei in adrena-lina (vezi mai jos). S-adenozil-homocisteina re-zultata in etapa 2, se scindeaza hidrolitic in homocisteina (hCys) si adenozina, hCys putand regenera metionina cu participarea N5-metil-tetrahidrofolat, donor de grupare metil. Cistatio-nina rezultata prin condensarea hCys cu Ser produce prin hidroliza a-cetobutirat si Cys. A-ceasta etapa (6) are un mecanism mai complex, nereprezentat in figura, implicand homoserina (hSer) si Cys, hSer conducand apoi la a-cetobu-tirat. Prin decarboxilarea oxidativa a acestuia re-zulta propionil-CoA si apoi, prin reactiile aratate mai sus, rezulta succinil-CoA.
Figura 16. Ciclul metilului activat. Numerele 1-6 indica enzimele din figura 17. 5-MeTHF= N5-metil-tetrahidrofolat; THF= acid tetrahidrofolic.
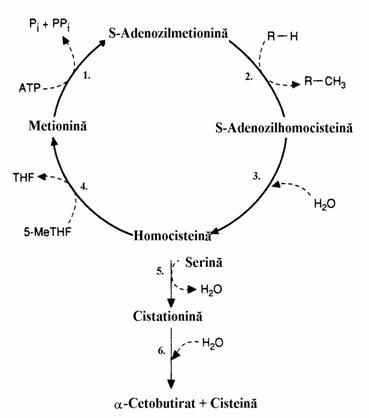
Se vede pozitia nodala ocupata de hCys si importanta Met in bilantul atomilor de sulf ceea ce determina caracterul lui de aminoacid esential. Importanta SAM ca donor de grupare metil se impune si in procesarea proteinelor chemotactice (chemoreceptori). In anii 1960 Julius Adler a studiat bazele moleculare ale che-motaxiei bacteriene. Aceasta incepe cu detectia unor chimicale prin intermediul chemoreceptorilor, prote-ine integrale sau periferice ale membranei plasmatice, unele pentru atractia, altele pentru respingerea in sensul gradientului chimic. Bacteriile inoata in mediul de suspensie prin rotatia filamentelor cu care sunt inzestrate pe suprafata. Schimbarea de directie a mis-carii lor este controlata prin intermediul proteinelor chemotactice acceptoare de metil (MCP). Acesti chemoreceptori transmit semnalul chemotactic prin membrana celulei bacteriene. MCP au centre de metilare in domeniul citosolic al lor continand mai multe resturi de Glu. Metilarea radicalilor Glu de catre SAM sub actiunea catalitica a unei metil-transferaze este intensificata de chimicalele atragatoare si inceti-nita de cele respingatoare. Acesta este semnalul prin care bacteria isi schimba directia in sensul gradientu-lui de concentratiei a substantei chimice atragatoare (glucoza de exemplu).
Intreaga schema reprezentata in figura 17 se refera la procesul numit trans-sulfurare.
Figura 17. Calea de degradare a Met cu formare de Cys si succinil-CoA. Enzimele implicate sunt: metio-nin adenozil transferaza (1), metilaza (2), adenozil homocisteinaza (3), homocistein metiltransferaza (vit. B12 dependenta) (4), cistationin b-sintaza (PLP-de-pendenta) (5), cistationin g-liaza (PLP-dependenta) (6), a-cetoacid dehidrogenaza (7), propionil-CoA carboxilaza (biotin-dependenta) (8), metilmalonil-CoA racemaza (9) si metilmalonil-CoA mutaza (coen-zima B12-dependenta) (10).
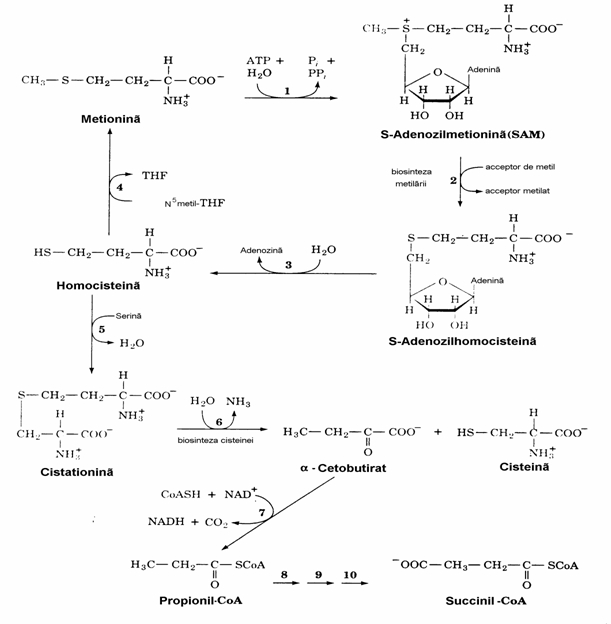
Din punct de vedere patologic, deficitul conge-nital al oricarei enzime implicate in trans-sulfu-rare determina acumularea unor aminoacizi care contin sulf. In hipermetioninemie este deficita-ra metionin adenoziltransferaza fara o simpto-matologie grava. In cistationurie este deficitara cistationin g-liaza cu acumulari de cistationina in sange si urina. Exista 4 tipuri de homocistei-nurii, primul tip datorandu-se deficitului de cistationin b-sintazei. Celelalte sunt cauzate de deficite ale enzimelor avand derivati ai acidului tetrahidrofolic drept substraturi (fig. 16, etapa 4). Nivele sanguine crescute de hCys se asociaza cu alterarea peretului vascular si cu deficitul de metilare - dezechilibru resimtit si in metabolis-mul acizilor nucleici.
1.3.2.5.Catabolismul aminoacizilor
cu catena ramificata.
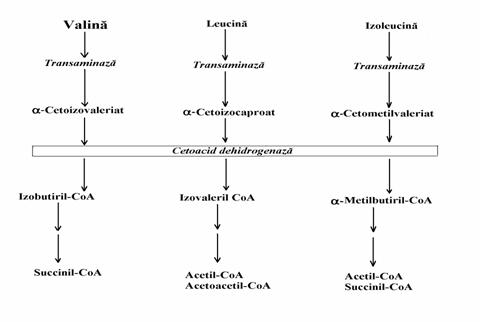
Val si Ile, aminoacizi cu catena ramifi-cata, produc succinil-CoA (fig. 18). Producand doar succinil-CoA valina este un aminoacid glu-coplastic. Izoleucina este un aminoacid mixt deoarece, ea produce atat succinil-CoA cat si CH3COSCoA din care se sintetizeaza corpi cetonici. Leucina este aminoacid cetoplastic deoarece prin catabolizare ea produce numai acetoacetat, corp cetonic.
Figura 18. Degradarea aminoacizilor cu catena ramificata (schema).
In figura 19 apar detaliat caile de degradare a aminoacizilor cu catena ramificata. Primele 3 reactii, transaminarea (1), decarboxilarea oxidati-va a a-cetoacizilor rezultati (2) si dehidro-genarea FAD-dependenta (3) sunt de acelasi tip. Mai mult, exista o singura enzima pentru transaminarea tuturor celor 3 aminoacizi. In ce priveste etapa decarboxilarii oxidative, comple-xele enzimatice au structuri similare cu ale complexului Pyr-DH folosind TPP, acidul lipoic si FAD drept coenzime ale enzimelor a-cetoacid DH, transacilaze si dihidrolipoil DH corespun-zatoare. In plus aceste complexe sunt inactivate, ca si in cazul Pyr-DH, prin fosforilare cu ATP de catre o protein kinaza. O fosfoprotein fosfataza defosforileaza aceste complexe enzimatice si, prin concurenta celor doua procese (fosforilare-defosforilare), este reglat catabolismul aminoacizilor cu catena ramificata. Inhibitorii protein kinazei sunt ADP, a-cetoacizii produsi, agenti hipolipidemici (clofibrat, dicloroacetat). Clofibratul este unul din primele medicamente antihipercolesterolemice. Cel mai puternic inhi-bitor dintre a-cetoacizi este a-cetoizocaproatul. In ce priveste dehidrogenarea FAD-dependenta a celor 3 aminoacizi, exista 2 enzime care catali-zeaza aceasta reactie, intre care izovaleril-CoA DH, printr-un mecanism similar dehidrogenarii FAD-dependente a acizilor grasi activati cu coenzima A, in cadrul b-oxidarii. In continuare caile de catabolizare ale Ile, Val si Leu se des-part. Inainte de a trece la prezentarea cailor de metabolizare a fiecarui aminoacid, este de mentionat similitudinea unor etape ale acestora.
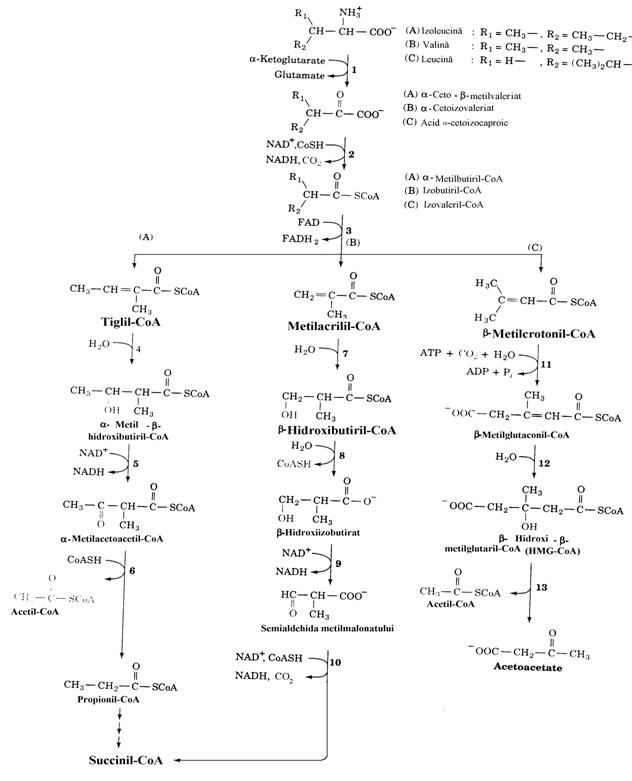
Figura 19. Caile de degradare ale aminoacizilor cu catena
ramificata: izoleucina (A), valina (B) si leucina (C). Primele 3
reactii ale fiecarei cai utilizeaza enzimele comune:
aminoacid cu catena ramificata aminotransferaza (PLP-dependenta)(1),
a-cetoacid
dehidrogenaza (2) si acil-CoA dehidrogenaza (3). Degradarea Ile
continua cu implicarea enzimelor: enoil-CoA hidrataza (4), b-hidroxiacil-CoA dehidro-genaza (5) si
acetil-CoA acetil transferaza (6). Degradarea Val implica: enoil-CoA
hidrataza (7), b-hidroxiizobutiril-CoA
hidrolaza (8), b-hidroxiizobuti-rat
dehidrogenaza (9) si metilmalonat semi-aldehida dehidrogenaza (10).
Degradarea Leu implica: b-metilcrotonil-CoA
carboxilaza (biotin-dependenta) (11), b-metilglutaconil-CoA hidrataza(12) si b-HMG-CoA-liaza(13).
In ce priveste catabolismul izoleucinei, este de mentionat ca etapele (3), (4), (5) si (6) coincid d.p.d.v. al tipurilor si secventei reactiilor cu b-oxidarea acizilor grasi. Spre deosebire de Val si Leu, Ile este catabolizata in continuare in 3 etape specifice (hidratarea, dehidrogenarea NAD+-de-pendenta si tioliza). Ultima etapa determina, prin produsii ei, caracterul mixt al acestui aminoacid. Un aspect interesant este identificarea allo-izoleucinei (allo-Ile) in plasma sanguina si urina umana deoarece avand 2 centre chirale Ile are 22 = 4 stereoizomeri prezentand fenomenul de dia-stereoizomerie. Racemizarea a-ceto-b-metil-va-leriatului, rezultat prin transaminarea L(+)-Ile explica formarea in vivo a allo-Ile. Caile de catabolizare a celor doi diastereoizomeri difera (calea S respectiv R). In sistemul (RS) L(+)-Ile este stereoizomerul (2S,3S) iar L(+)allo-Ile este stereoizomerul (2S,3R). Principala cale de catabolizare a lui este calea S.
In ce priveste valina secventa catabolica contine 4 etape specifice. Semialdehida metil-malonica sufera in etapa (10) o transformare mai complexa pentru a se transforma in succinil-CoA (decarboxilare oxidativa cu formare de metil-malonil-CoA si apoi conversia acestuia in succi-nil-CoA sub actiunea mutazei (fig. 15). O cale de transformare mai putin intensa este transaminarea semialdehidei cu formare de b-aminoizobutirat care este excretat in mod normal in urina (trans-formare neprezentata in figura 19).
Leucina parcurge 3 etape specifice: car-boxilare, hidratare si clivajul b-HMG-CoA. Car-boxilarea este reactia care determina caracterul cetoplastic al Leu. b-HMG-CoA este un metabo-lit cunoscut in cetogeneza si colesterologeneza.
Din punct de vedere patologic se cunosc tulburari congenitale ale tuturor etapelor de catabolizare prezentate in figura 19. Cea mai importanta este boala cu urina cu miros de artar (Maple Syrup Urine Disease) cu o incidenta de 1/100000 de nou-nascuti. Urina si sangele acestor bolnavi contin mari cantitati de Ile, Val si Leu precum si de a-cetoacizii corespunzatori. Boala este evidenta la sfarsitul primei saptamani de viata extrauterina. Nou-nascutii se alimenteaza cu greu, vomita si sunt letargici. Diagnosticul poate fi pus in prima saptamana dupa nastere prin analiza enzimatica. Copiii care supravietuiesc sufera puternice tulburari neurologice si moartea intervine dupa primul an de viata. Deficitul enzimatic care sta la baza acestei boli este lipsa sau activitatea redusa a complexului a-cetoacid dehidrogenaza. Diag-nosticul timpuriu si instituirea unei diete terapeutice, in care proteinele sunt inlocuite cu amestecuri de aminoacizi din care lipsesc cei 3 aminoacizi cu catene ramificate, previn simpto-mele aratate. Cand nivelele plasmatice al Ile, Val si Leu se normalizeaza copiii pot fi alaptati sau alimentati cu cantitati adecvate de alimente care contin proteine in compozitia carora intra cei 3 aminoacizi esentiali. In cetonuria intermitenta cu catene ramificate deficitul enzimatic este acelasi ca si in cazul anterior, insa mai putin sever, simptomele aratate aparand cu intermi-tenta. Acidemia izovalerica este o boala conge-nitala cu deficit de izovaleril-CoA-dehidroge-naza (FAD-dependenta). Bolnavii expira un miros de "branza", vomita, au acidoza si intra in coma daca ingera mari cantitati de proteine. Hidroliza izovaleril-CoA acumulate produce acid izovaleric care apare in urina si sudoare.
Hipervalinemia este o boala metabolica rara in care nivelul plasmatic al Val este ridicat (nu si cele ale Ile si Leu) datorita incapacitatii de transaminare a valinei. Faptul ca etapa transami-narii (reactia 1 in fig. 19) este catalizata de o singura enzima sugereaza afinitati diferite ale celor 3 aminoacizi fata de transaminaza.
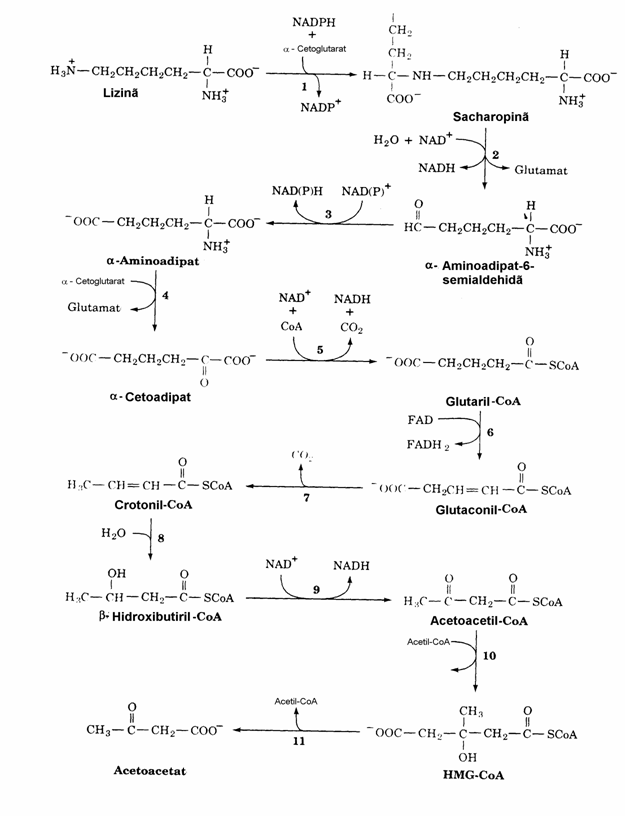
Ca si Leu, Lys este un aminoacid ceto-plastic produsii sai fiind acetoacetatul si CH3COSCoA. Desi exista mai multe cai de cata-bolizare a Lys, cea care are drept intermediar sacharopina predomina in ficatul mamiferelor (fig. 20). Aceasta preponderenta explica faptul ca in defectul enzimatic al sacharopin dehidroge-nazei NADPH-dependente (reactia 1 in figura) se produce hiperlizinemie si hiperlizinurie cu simptome de retardare mintala si fizica. De fapt exista doua forme ale acestei tulburari: hiper-lizinemia periodica asociata cu hiperamone-mie. Hiperamonemia se datoreaza inhibitiei com-petitive pe care Lys in exces o exercita asupra arginazei hepatice. Aceasta inhibitie incetineste ciclul ureogenetic determinand acumularea amo-niacului. O alta forma este hiperlizinemia per-sistenta fara hiperamonemie. Este de remarcat ca ultimele doua etape ale catabolizarii Lys (reactiile 10 si 11, fig. 20) coincid cu doua etape ale cetogenezei, relevand inca o data rolul de me-tabolit central al b-HMG-CoA, ca si cel al altor compusi macroergici cum sunt CH3COSCoA si izopentenil-PP.
Figura 20. Calea de degradare a Lys in ficatul mamiferelor. Enzimele implicate sunt: sacharopin dehidrogenaza (NADPH-dependenta) (1), sacharopin dehidrogenaza (NAD+-dependenta) (2), aminoadipat semialdehida dehidrogenaza (3), aminoadipat amino-transferaza (PLP-dependenta) (4), a-cetoacid dehi-drogenaza (5), glutaril-CoA dehidrogenaza (6). decarboxilaza (7), enoil-CoA hidrataza (8), b-hidroxi-acil-CoA dehidrogenaza (9), b-HMG-CoA sintaza (10) si b-HMG-CoA liaza.
Prima transformare a fenilalaninei este hidroxilarea ei in pozitia para (fig. 21), singura reactie ce, pe deoparte o transforma in alt aminoacid "standard", Tyr, iar pe de alta parte incepe calea de degradare a scheletului de atomi de carbon ai Phe. Producand un intermediar krebsian si un corp cetonic, Phe si Tyr, apartin grupei aminoacizilor micsti. Conversia Phe Tyr necesita fenilalanin hidroxilaza al carei cofactor este 5,6,7,8-tetrahidrobiopterina (fig. 22). Biopte-rina este un derivat de pteridina, al carei nucleu il regasim in structura acidului folic.
Forma total
redusa a biopterinei rezulta prin reducerea 7,8-dihidrobiopterinei cu
NADPH de catre dihidrofolat
reductaza, sau cu NADH de catre dihidropteridin
reductaza. Fenilalanin hidroxilaza este o monooxigenaza, adica o oxi-daza cu functiune
mixta, al carei substrat princi-pal este Phe iar cosubstratul este
tetrahidrobio-pterina. Denumirea de monooxigenaza este data atunci
cand, in reactie, un atom din molecula de O2 apare in produsul
hidroxilat iar celalalt este incorporat intr-o molecula de apa.
Hidroxilaze (monooxigenaze) am mai intalnit in reactiile de metabolizare a
colesterolului.
Transaminarea a-cetoglutarat-depen-denta a Tyr produce p-hidroxifenilpiruvat, ea fiind catalizata de o transaminaza hepatica in-ductibila (reglabila prin inductie-represie enzi-matica).
Conversia p-hidroxifenilpiruvatului in homogen-tizinat este catalizata de o dioxigenaza ce contine ioni Cu2+. Denumirea de dioxigenaza se datoreaza faptului ca in reactie ambii atomi ai moleculei O2 sunt incorporati in produs, ca si in cazul Trp dioxigenazei (fig. 23). Agentul reducator implicat in aceasta reactie este acidul ascorbic. Din aceasta cauza bolnavii de scorbut excreta p-hidroxifenilbutirat in urina. In continu-are, o alta dioxigenaza "sparge" acidul homo-gentizinic in 4-maleilacetoacetat, compus nearo-matic. De fapt, aproape toate reactiile de clivaj a nucleului aromatic din sistemele biologice sunt catalizate de dioxigenaze, o clasa de enzime descoperita de Osamu Hayaishi. Enzima, prezen-ta in ficatul mamiferelor, contine ioni Fe2+. In continuare, transformarea 4-maleil-acetoacetatu-lui in 4-fumarilacetoacetat are un mecanism mai complex (neprezentat in figura) in care intervine glutationul. El hidrogeneaza scheletul acidului maleic, dehidrogenarea ulterioara a acestuia, sub actiunea complexului enzimatic maleil-aceto-acetat cis trans izomeraza, producand scheletul acidului fumaric. Fumaril-acetoacetat hidrola-za catalizeaza reactia finala de clivaj cu produce-re de fumarat si acetoacetat.
Deficitele congenitale ale enzimelor caii de catabolizare a Phe determina boli metabolice inascute care au fost dintre cele mai amanuntit studiate. Alcaptonuria, o tulburare inascuta a acestei cai, a fost semnalata (simptomatic) de Zacutus Lusitanus in 1649 care in limbajul arhaic al vremii sale descria pacientul, un baiat a carui urina emisa se inegrea dupa expunerea la aer. Bolnavul a fost supus unui tratament complex incluzand sangerari, purgatii, bai, dieta hidratanta si mai multe medicamente. Nici unul din aceste tratamente n-a avut vreun efect asupra simp-tomului mentionat dar nici necazurile prezise pacientului nu s-au confirmat. El s-a casatorit, a avut o familie numeroasa si o viata lunga, singura deosebire de semeni fiind urina sa care dupa emisie se inegrea cu timpul pana la culoarea cafelei. Abia in 1902 Archibald Garrod a aratat ca alcaptonuria este o boala autosomala recesiva de tip Mendelian. El a prevazut in 1908 intr-un articol aparut in prestigioasa revista "Lancet" etiopatogenia ei ca fiind deficitul unei enzime implicate in degradarea Phe. Aceasta a fost prima boala careia i s-a descoperit acest determinism, ceea ce a constituit o piatra de hotar in intele-gerea bolilor metabolice. Cu alte cuvinte Sir A. Garrod a fost primul care a inteles relatia directa dintre gene si enzime idee dezvoltata in celebra sa carte "Erorile inascute ale metabolismului".
Azi este clar ca alcaptonuria este deter-minata de deficitul congenital al homogentizi-nat dioxigenazei. In conditii normale acidul homogentizinic (alcaptonul) se gaseste in canti-tati infime in plasma sanguina si urina. In alcap-tonurie, numita si acidurie homogentizinica, nivelul plasmatic este nesemnificativ in schimb cel renal este mare, ceea ce se datoreaza unei viteze de clearance renal care depaseste viteza filtrarii glomerulare. Faptul se datoreaza excretiei homogentizinatului in lumenul tubular renal. Pre-zenta acestui acid in urina se poate evidentia prin mai multe metode intre care simpla alcalinizare (cuvantul "alkapton" inseamna alcalii in limba araba) sau reactia cu FeCl3 care produce o culoare purpuriu-inchisa.
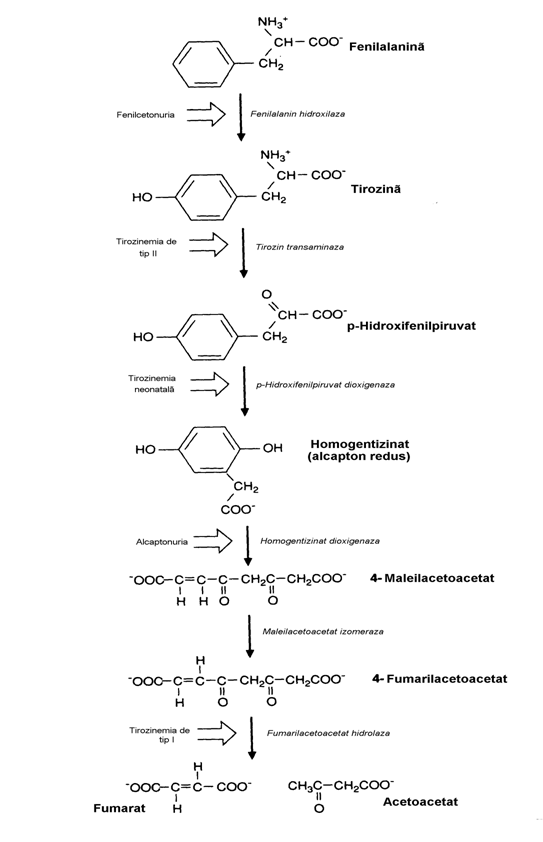
Figura 21. Calea de degradare a Phe. Sunt indicate si tulburarile
metabolice cu precizarea enzimelor defici-tare corespunzatoare. Transformarea
Tyr in compusul ceto-corespunzator are loc prin transaminare cu a-cetoglutarat.
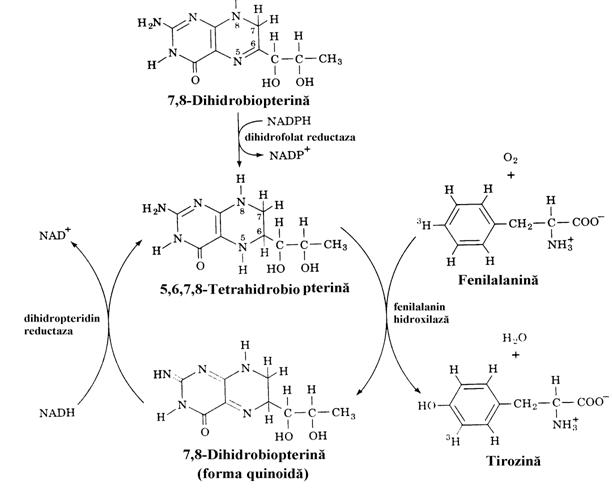
Figura 22. Mecanismul reactiei de hidroxilare a fenil-alaninei(fig. 21).
Fenilalanin hidroxilaza al carei cosubstrat reducator este
tetrahidrobiopterina care se regenereaza pe doua cai folosind ca
donatori de hidrogen NADPH si NAD.
Ocronoza (inchiderea la culoare a unor tesuturi) apare mult timp dupa cresterea nivelului homogentizinuriei, ceea ce sugereaza o infiltratie lenta a tesuturilor conjunctive. Cu timpul apare si o forma de artrita. Mecanismul ocronozei impli-ca, ca si in alcaptonurie, oxidarea acidului homo-gentizinie de catre polifenol oxidaza cu formare de benzochinonacetat, forma chinonica (oxidata) a alcaptonului care se polimerizeaza si se aso-ciaza la macromoleculele tesutului conjunctiv.
Din punct de vedere terapeutic nu exista un tratament eficient, dupa instalarea ocronozei. Este de remarcat faptul ca, datorita existentei instalatiilor sanitare moderne de evacuare rapida a urinei, bolnavii nu observa simptomul carac-teristic, inegrirea in timp, in contact cu aerul a acesteia. Pe de alta parte, diagnosticul timpuriu al alcaptonuriei si impunerea unei diete saracite in Phe si Tyr poate preveni alcaptonuria.
Fenilcetonuria clasica (oligofrenia fe-nilpiruvica, hiperfenilalaninemia de tip I)
are, spre deosebire de alcaptonurie, efecte devas-tatoare in special prin retardarea mintala severa. Alte semne clinice sunt psihoze, eczeme, miros de "soarece". Greutatea creierului la acesti bolnavi este sub-normala, gradul de mielinizare a nervilor este defectuos si fenilcetonuricii netra-tati au o speranta de viata redusa (20-30 ani). Cauza este absenta sau deficitul de fenil-alanin hidroxilaza. Frecventa este 1:10000 de nou-nascuti, 1% din pacientii institutionalizati avand acest defect. Phe hidroxilaza hepatica are doar ¼ din activitatea ei normala ceea ce se soldeaza cu cresterea nivelului sanguin al Phe. Din aceasta cauza are loc o "diversiune metabolica" in ficat, adica Phe este nevoita sa parcurga o cale mai putin folosita. Prin deaminare se formeaza fenil-piruvat care prin reducere se transforma in fenillactat. Decarboxilarea si oxidarea fenil-piruvatului da nastere la fenilacetat. Ultimul metabolit este excretat in urina in stare conden-sata cu Gln, fenil-acetil-glutamina. Fenilceton-uricii par a fi normali la nastere simptomele bolii devenind evidente la varsta de 1 an.
Fenilcetonuria clasica a fost prima boala inascuta (umana) a metabolismului al carei defect enzima-tic a fost identificat (in 1947). Prognosticul sever si necesitatea punerii in evidenta de timpuriu a oligofreniei fenilpiruvice, face necesara analiza prenatala a ADN. ADN-ul din celulele prezente in mai putin de 10ml de lichid amniotic sau din probele biotice de villus chorionic poate fi anali-zat prin tehnica de transfer Southern. In unele boli metabolice congenitale cu simptomatologie grava diagnosticul precoce se poate pune, dease-menea, prin analiza enzimatica a lichidului amni-otic numita amniocenteza.
Terapia genica (clonarea unei gene care codifica enzima sanatoasa, menita sa inlocuiasca enzima deficitara a bolnavului), aflata inca la inceputurile ei experimentale, da sperante in ce priveste inlaturarea bolii metabolice inca in cur-sul vietii pacientului. In prezent, deteriorarea performantei mintale a bolnavilor cu fenilceto-nurie clasica poate fi prevenita prin aplicarea unei diete speciale, avand un continut mic de Phe, in primii 5-10 ani de viata, dupa care, revenirea la o dieta obisnuita nu mai produce "injurii" siste-mului nervos central chiar daca fenilalaninemia creste peste nivelul sanguin normal. Intr-un studiu pe un grup de fenil-cetonurici, la care dieta a fost instituita imediat dupa nastere, media IQ s-a mentinut in jurul valorii 93, fata de un grup martor de fenilcetonurici, frati cu cei din primul grup, la care dieta s-a instituit abia dupa un an de la nastere si la care media IQ a atins valoarea 53. Identificarea fenilpiruvatului in urina nou-nascu-tilor se poate face simplu cu FeCl3 cand apare o coloratie verde-maslinie. Totusi, determi-narea in timp a fenilalaninemiei este un test mai sigur pentru decelarea bolii datorita rezultatelor fals-pozitive determinate de intarzierea in maturizare a enzimelor catabolizarii Phe.
Hiperfenilalaninemiile de tip II si III sunt deter-minate de defecte ale reducerii dihidrobiopte-rinei (fig. 22). Cele de tip IV si V se datoreaza defectelor de biosinteza a dihidrobiopterinei.
Exista cateva boli cu hipertirozinemie. In tirozinemia de tip I etiopatogenia este mai com-plexa fiind implicate deficite multiple in special al enzimei fenilacetoacetat hidrolaza.
In tirozinemia de tip II (Richner-Han-hart) enzima deficitara este tirozin transamina-za. Nivelul Tyr la nou-nascuti este ridicat (4-5mg% in loc de 1,3mg%) si apar leziuni ale ochiului, pielii si retardare mintala. In tirozi-nemia neonatala enzima deficitara este p-hidro-xifenilpiruvat dioxigenaza. Nivelele sanguine ale Phe si Tyr sunt ridicate. Terapia se bazeaza pe dieta saraca in proteine.
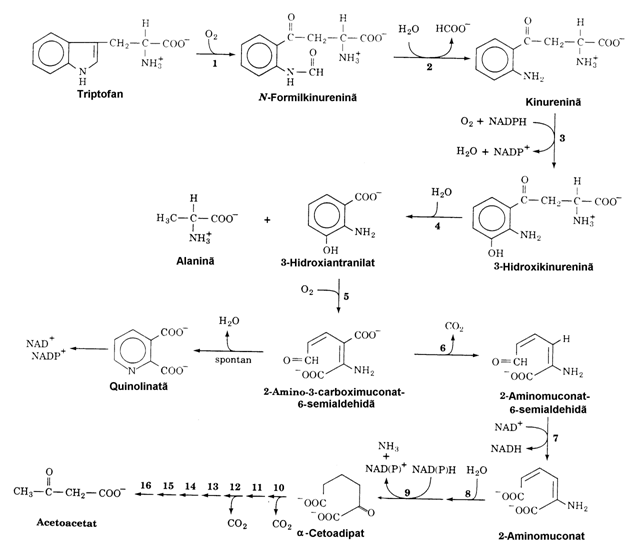
Cum se vede in figura 23 si triptofanul produce acetoacetat prin catabolizare. Totusi, el intra in categoria aminoacizilor micsti deoarece in etapa 4 rezulta Ala care este precursor gluco-neogenetic. Atat atomii de carbon ai catenei laterale (Ala) cat si cei ai nucleului aromatic pirolic pot fi incorporati in intermediari amfi-bolici pe calea kinurenina-antranilat importan-ta atat pentru degradarea Trp cat si pentru conver-sia lui in nicotinamida(vezi vol. I).
Din punct de vedere patologic, deficitul de vitamina B6 altereaza activitatea kinure-ninazei (enzima PLP-dependenta) si determina acumularea kinureninei care, ajungand in tesutu-rile extrahepatice se transforma intr-un metabolit anormal, xanturenat care ajunge in urina. Cum am vazut in vol.I, PLP este necesar si conversiei Trp NAD+. De aceea, deficitul de vitamina B6, determina la om pe langa excretia xanturenatului si un deficit de NAD+ si NADP+. Boala Hartnup este o anomalie ereditara de metabolizare a Trp cu simptome pelagroase, ataxie cerebrala inter-mitenta si retardare mintala. Urina contine canti-tati crescute de Trp si indol-acetat. Cum vom vedea triptofanul se transforma in neuromedia-torul serotonina.
Figura 23. Calea de degradare a Trp. Enzimele impli-cate sunt: triptofan 2,3 dioxigenaza (1), formamidaza (2), kinurenin-3-monooxigenaza (3), kinureninaza (PLP-dependenta) (4), 3-hidroxi-antranilat-3,4-dioxi-genaza (5), amino carboximuconat semialdehida decarboxilaza (6), aminomuconat semialdehida dehi-drogenaza (7), hidrataza (8), dehidrogenaza (9). Enzimele 10-16 coincid cu enzimele care catalizeaza degradarea Lys in reactiile 5-11 (fig. 20).
1.3.2.8.
Conversia aminoacizilor aro-matici in neuromediatori si alti
compusi
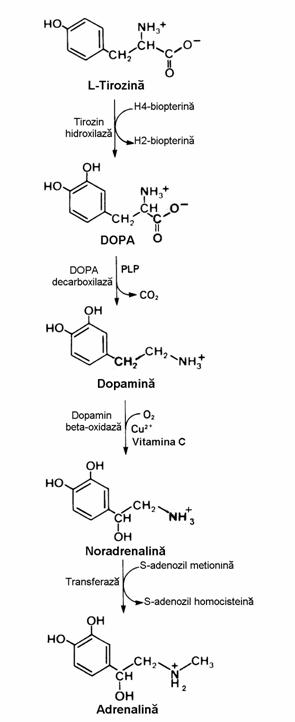
In figura 24 se vede ca tirozina este precursor al catecholaminelor - noradrenalina si adrenalina care se formeaza in neuroni respectiv celule cromafine. Prima reactie, conversia Tyr → DOPA are acelasi mecanism cu al conversiei Phe →Tyr(fig. 22). Dihidroxifenil alanina(DOPA) este intermediar de pornire al unor bifurcatii metabolice pentru sinteza melaninelor in melano-cite (celule producatoare de pigmenti). Tirozin hidroxilaza neuronala este diferita de cea din melanocite, in pofida identitatii substraturilor si a produsilor. Deficitul congenital de hidroxilaza melanocitara determina boala numita albinism al carei principal simptom este lipsa de pigmentatie, bolnavii sunt albinoizi.In continuare prin decarboxilare si "β-oxidare" rezulta noradrenali-na (norepinefrina). Ultima enzima are drept co-factori cuprul si vitamina C. In celulele croma-fine medulosuprarenale exista o enzima (fenil-etanolamin-N-metil-transferaza) care utilizeaza SAM pentru conversia noradrenalina → adrenalina. O tumora a aceste celule determina supraproductia de adrenalina in boala numita feo-cromocitom printre ale carei simptome se distin-ge hipertensiunea arteriala.
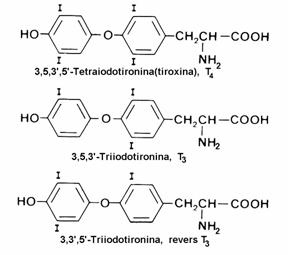
Tirozina este precursor al hormonilor tiroidieni - triiodotironina(T3), revers-T3 si tetra-iodotironina(tiroxina), care se sintetizeaza prin organificarea ionilor iodura in celulele foliculilor tiroidieni. Supraproductia sau insuficienta aces-tor hormoni determina hiper- respectiv hipoti-roidismul
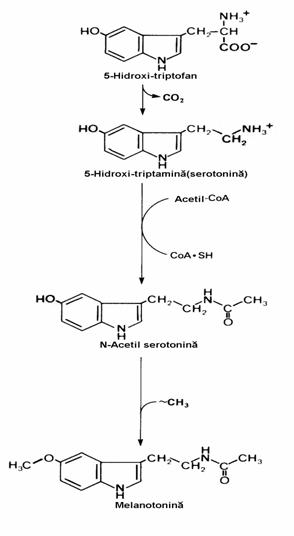
Triptofanul este precursor al serotoniei si melatoninei(fig. 24). Formarea 5-hidroxi-tripto-fanului necesita acelasi tip de echipament enzi-matic ca si conversia conversiei Phe →Tyr. Mai mult, fenilalanin hidroxilaza hepatica catalizeaza si oxidarea triptofanului. In continuare, decarbo-xilarea 5-HO-Trp produce serotonina, un puternic vasoconstrictor si stimulator al contractiei mus-chilor netezi. Decarboxilaza se gaseste in ficat si stomac. Cea mai mare parte a serotoninei este metabolizata prin deaminare oxidativa sub actiu-nea monoamin oxidazei(MAO). Inhibitorii acestei enzime constituie o clasa de medicamente antidepresive, alaturi de inhibitorii selectivi ai
recaptarii serotoniei (fluoxetin), antidepresivele policiclice (imipramina) si sarurile de litiu.
Figura 24 Sinteza catecholaminelor si a serotoninei din aminoacizi aromatici. Hormoni tiroidieni.
Copyright © 2024 - Toate drepturile rezervate